Revisión anual
El síndrome del QT corto congénito: avances en los últimos años
Andrés R Pérez-Riera, Luiz C De Abreu, Raimundo Barbosa-Barros, Adail Paixão-Almeida
Revista del Consejo Argentino de Residentes de CardiologÃa 2016;(135): 0141-0147
Este articulo contiene material suplementario excusivo webEl síndrome de QT corto (SQTC) congénito es una entidad hereditaria, de transmisión autosómica dominante o esporádica, heterogénea genotípica y fenotípicamente, huérfana o rara, sin cardiopatía estructural aparente. Su característica principal es la presencia en el electrocardiograma (ECG) de intervalos QT/QTc cortos (entre 315 y 360 milisegundos), causado por mutaciones genéticas que afectan los canales de potasio (K+) y de calcio (Ca2+).
Se describen tres variantes principales: el SQTC tipo 1, que es ocasionado por una mutación en el gen HERG, (o KCNNH2 en la nueva denominación) y afecta el canal rectificador de salida rápida de K+ activado por voltaje (IKr); el tipo 2, donde se halló una alteración en el gen KCNQ1, que controla la corriente de salida lenta rectificadora de K+ (IKs) en fase 3; y el tipo 3 en el que se identificó una mutación en el gen KCNJ2, que codifica la proteína del canal rectificador de entrada de K+, causante de un aumento significativo en la corriente de entrada (Ik1). Estas tres variantes junto al SQTC 7, que compromete el canal de salida inicial de K+ o canal Ito de la fase 1, constituyen las formas puras, pero también se mencionan alteraciones del canal de Ca2+ (SQTC tipos 4, 5 y 6), que se catalogan como superposiciones fenotípicas de canalopatías u overlapping.
La entidad se manifiesta clínicamente por palpitaciones, mareos, síncope y elevada tendencia a la muerte súbita cardíaca, por eventos de fibrilación auricular y taquicardia ventricular polimórfica/fibrilación ventricular.
El cardiodesfibrilador implantable es la terapia de primera línea, y el tratamiento farmacológico puede estar indicado en algunos casos.
Palabras clave: canalopatÃas, sÃndrome de QT corto, fibrilación auricular, muerte cardiaca súbita.
Short QT syndrome (QTC) is an orphan, congenital or hereditary entity, with autosomal dominant or sporadic transmission, genotypically and phenotypically heterogeneous, with no apparent structural heart disease. Its main feature is the presence in the electrocardiogram (ECG) of short QT / QTc (between 315 and 360 milliseconds according to the author), caused by genetic mutations that affect potassium (K +) and calcium (Ca2 +) channels.
Three main variants are described: the SQTC type 1, which is caused by a mutation in the HERG gene (or KCNNH2 in the new nomenclature) and affects the fast rectifier output voltage-activated K+ channel (IKr); type 2, where there is a KCNQ1 alteration that controls the slow rectifier K+ current in phase 3; and type 3 in which there is a mutation in the KCNJ2 gene, that encodes the inward rectifier K+ channel protein, causing a significant increase in the inward current (Ik1). These three variants and SQTC7, which affects the initial outward K+ channel, or Ito channel from phase 1, constitute pure forms, but alterations of Ca2+ channel (SQTC types 4, 5 and 6) are also mentioned, classified as phenotypic overlapping channelopathies.
The entity manifests clinically by palpitations, dizziness, syncope and high tendency to sudden cardiac death, by events of atrial fibrillation and polymorphic ventricular tachycardia / ventricular fibrillation.
The implantable cardioverter defibrillator is the first-line therapy, and drug therapy may be indicated in some cases.
Keywords: channelopathies, short QT syndrome, atrial fibrillation, sudden cardiac death.
Los autores declaran no poseer conflictos de intereses.
Fuente de información Consejo Argentino de Residentes de Cardiología. Para solicitudes de reimpresión a Revista del CONAREC hacer click aquí.
Recibido 2015-12-20 | Aceptado 2016-01-05 | Publicado 2016-09-01

Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial-SinDerivar 4.0 Internacional.








Introducción
El síndrome de QT corto (SQTC) congénito es una entidad hereditaria, de transmisión autosómica dominante o esporádica, heterogénea genotípica y fenotípicamente, huérfana o rara (afecta ≤1 de cada 1.500 personas), sin cardiopatía estructural aparente. Es integrante de los así llamados defectos de los canales iónicos, canalopatías cardíacas, o enfermedades eléctricas primarias1 y su característica principal es la presencia en el electrocardiograma (ECG) de intervalos QT/QTc extremadamente cortos, principalmente en las formas causadas por mutaciones genéticas que afectan los canales de potasio (K+), y un poco menos cortos en las formas ocasionadas por mutaciones que afectan el canal de calcio (Ca2+).
No existe consenso sobre considerar o no como parte del SQTC las ocasionadas por mutaciones de Ca2+ descriptas por Antzelevitch2. Estas formas, en realidad, constituyen superposiciones fenotípicas de canalopatías (overlapping en la literatura anglosajona) y no formas puras de SQTC.
No existe un valor de corte diagnóstico universalmente aceptado del QTc para considerarlo exclusivo de esta rara canalopatía. Giustetto ha propuesto un valor de QTc≤340-360 milisegundos (ms)3. Spears y Gollob dan un valor de corte 10 ms menor (QTc< 330 ms)4. Valores del QTc de 340-350 ms son considerados normales por algunos autores, habiendo sido observados sin cualquier aumento del riesgo muerte cardíaca súbita (MCS)5. Quienes suscriben consideran sospechosos valores de QTc≤360 ms.
Otra característica de destaque del SQTC es su elevada tendencia a la aparición de episodios de fibrilación auricular (FA) paroxísticos. Este tipo de FA se caracteriza por ser desencadenado por una extrasístole auricular, tener inicio y fin abruptos, y presentarse con carácter de episodios recurrentes (2 o más episodios de FA con duración >30 segundos [s]), autolimitados, por un período generalmente < 24 horas (hs), aunque pueden alcanzar las 48 hs o más.
La presencia de FA en niños, adolescentes y/o adultos jóvenes debe hacernos sospechar la presencia de SQTC, sobre todo cuando el intervalo QTc sea corto6, antes de rotularla como FA aislada, idiopática, primaria o criptogénica (lone atrial fibrillation). Esta se define como toda FA donde no exista cardiopatía estructural, hipertensión, preexcitación ventricular o enfermedades extracardíacas predisponentes, como el hipertiroidismo, alcoholismo, uso de drogas, feocromocitoma, sensibilidad a adrenérgicos, y la denominada “vagalmente mediada” (una rara forma observada generalmente en hombres de mediana edad, habitualmente posprandial o en reposo). Tampoco pueden existir antecedentes familiares positivos para FA paroxística precoz o MCS. En todos estos casos, deben ser descartadas las numerosas causas genéticas de FA, dentro de las cuales está incluido el SQTC.
Otra característica fundamental y exclusiva del SQTC consiste en que el intervalo QTc no se modifica proporcionalmente con los cambios bruscos en la frecuencia cardíaca (FC)7. En comparación con la población normal, los pacientes portadores del SQTC tienen menos variación del intervalo QTc en relación con los cambios en la FC; esto da lugar a una interpretación errónea del intervalo QTc en presencia de una FC más rápida y el diagnóstico de falsos negativos. El método de Holter puede ser útil para esclarecer esta duda, ya que permite la medición del intervalo QTc durante un período de FC lenta, como ocurre en el sueño.
Frecuentemente las ondas T son de gran voltaje, puntiagudas8-10 y de bases estrechas, muy semejantes a las ondas T de la hiperkalemia. El bucle T del vectocardiograma es anormal en su voltaje y en el ángulo QRS/T11. No obstante, el bucle T permanece asimétrico, con la rama aferente de inscripción lenta y la eferente rápida, como ocurre normalmente9.
La entidad se manifiesta clínicamente por palpitaciones, mareos, síncope, elevada tendencia a la MCS, por eventos de FA y taquicardia ventricular polimórfica/fibrilación ventricular (TVP/FV).
Electrofisiológicamente, hay un constante acortamiento significativo de los períodos refractarios de las aurículas y ventrículos, siendo inducibles FA/FV sostenida por estimulación programada12,13.
Se han identificado unas pocas familias con varios tipos de mutaciones que afectan los canales de K+ y Ca2+. Hasta la fecha se han informado mutaciones en siete genes asociados con el SQTC: HERG o KCNH2 (SQTC1), KCNQ1 (SQTC2), KCNJ2 (SQTC3), CACNA1C (SQTC4), CACNB2b (SQTC5), CACNA2D1 (SQTC6) y caveolina-3 (SQTC7?) (familia de Brasil, todavía no publicada, pero con mutación identificada que afecta el canal de salida inicial de K+ o canal Ito de la fase 1 correspondiente al punto J del ECG de superficie).
Se categorizaron como SQTC1-SQTC7 en base a la cronología de su descubrimiento. La Tabla 1 muestra la proteína y subunidad involucrada, el canal afectado y la alteración funcional que ocasiona en él.
Las 4 mutaciones en los canales de K+ llamadas SQTC1(Ikr), SQTC2(Iks) SQTC3(Ik1) y SQTC7? tienen como contrapartida o imagen en espejo genética una variante del síndrome del QT prolongado (SQTL): SQTC2 con el SQTL1, el SQTC1 con el SQTL2, SQTC7? con el SQTL9 o síndrome de Andersen-Tawil, porque ejercen efectos opuestos de ganancia de función sobre las corrientes rectificadoras de salida rápida y lenta de K+ (IKr y IKs) y la corriente rectificadora cardíaca de entrada de K+ (IK1) en contraste con la pérdida de función de los canales de K+ de los SQTL (Figura 1). La Figura 2 muestra el ECG del primer caso de SQTC observado en Brasil y publicado por Pérez-Riera et al.9. Este caso estaba asociado a bloqueo de la rama derecha. La Figura 3 muestra la correlación ECG/VCG en los tres planos.
Características de las principales variantes del SQTC
I. Síndrome de QT corto congénito tipo 1 o fenotipo SQTC1 (IKr)
El SQTC1 (OMIM #609620) es causado por una mutación en el gen HERG (human ether-a-go-go related gene, en la nomenclatura italiana, o KCNNH2 en la nueva denominación). Este gen codifica la subunidad α formadora del poro del canal rectificador de salida rápida de K+ activado por voltaje (IKr). Esta mutación KCNNH2 fue identificada por primera vez en enero de 2004 por Ramón Brugada et al. cuando trabajaba en el Laboratorio Masónico14. Los autores identificaron dos mutaciones missense en dos familias, que resultaron en el mismo cambio de aminoácidos (N588K) en la región del bucle S5-P del gen HERG (KCNNH2), que controla el canal rectificador de salida rápido de K+ (IKr). Las mutaciones IKr aumentan dramáticamente la velocidad de salida de K+ en fase 3 (correspondiente a la onda T del ECG de superficie), lo que resulta en un acortamiento heterogéneo de la duración y la refractariedad del PA, y en una reducción de la afinidad de estos canales rápidos de K+ con los bloqueantes IKr como los antiarrítmicos de la clase III.
La ocurrencia de MCS en los primeros 12 meses de vida sugiere la posibilidad de un vínculo entre las mutaciones del gen KCNH2 de ganancia de función y el síndrome de muerte súbita infantil (SIDS: sudden infant death syndrome)15.
II. Síndrome de QT corto congénito fenotipo tipo 2 o SQTC2(IKs)
Bellocq et al. informaron en un hombre de 70 años, resucitado con éxito luego de un episodio de FV, que el intervalo QTc era muy corto (290 ms posevento) y permaneció próximo a este valor en 3 años de seguimiento. Su historia familiar era negativa, no refería síntomas previos ni se comprobó cardiopatía estructural. Los autores identificaron una mutación missense en el gen KCNQ1 (OMIM: #609621), el cual controla la corriente de salida lenta rectificadora de K+ (IKs) en fase 3 (correspondiente a la onda T del ECG de superficie), ocasionando ganancia en la función del canal. También identificaron algunos pocos casos esporádicos de esta variante16.
Maltret et al. observaron disfunción auditiva central en pacientes portadores de la variante SQTC217, lo que sugiere que pueda ser la imagen en espejo genética del SQTL recesivo de Jervell y Lange-Nielsen variante 1. Este síndrome afecta en forma opuesta el canal de salida lenta de K+ (disminución de la función), con sordera congénita, central, neurosensorial, bilateral y profunda. En esta entidad la mutación también ocurre en el mismo gen KVLQT1 (KCNQ1 del SQTC2), con locus en el cromosoma 11p15.5.
III. Síndrome de QT corto congénito fenotipo tipo 3 o SQTC3 (IK1)
Priori et al.18 identificaron una mutación missense en el gen KCNJ2. Estas mutaciones se observaron en una niña asintomática de 5 años que presentaba un intervalo QTc de 315 ms y ondas T de base estrecha y amplias, y en el padre de 35 años con un intervalo QTc corto (320 ms) además de historia de episodios presincopales y palpitaciones desde la adolescencia. Se identificó una mutación en el gen KCNJ2 causante de un aumento significativo en la corriente de entrada Ik1 (cardiac inwardly rectifying potassium current), el mismo canal afectado en la variante de SQTL conocida como síndrome de Andersen Tawil o SQTL7 (OMIM #170390), localizado en el cromosoma 17q24.
Hattori et al. identificaron una mutación M301K heterocigota en el gen KCNJ2 que ocasiona ganancia de función del canal rectificador de entrada de K+, asociada a SQTC319. Se ha descripto otra mutación (E299V) en el gen KCNJ2, que codifica la proteína del canal rectificador de entrada de K+ (Kir2.1). Se observaron cambios proarrítmicos en el PA con pérdida y ganancia de función en el canal IK1 (Kir2.2), en asociación con el síndrome de Andersen-Tawil tipo 1 (ATS OMIM #170390) (LQT7) y el SQTC3 respectivamente10.
Principales características del SQTC
-
Ausencia de cardiopatía estructural.
-
Síndrome clínico-electrocardiográfico.
-
Hereditario, autosómico dominante o esporádico y genotípica y fenotípicamente heterogéneo.
-
Frecuente historia familiar positiva para síncope y MCS en familiar de primer grado joven (< 50 años).
-
Manifestación de síncopes, MCS, palpitaciones, mareos y alta tendencia a la aparición de episodios de FA paroxísticas.
-
Intervalos QT y QTc extremadamente cortos, constantes y uniformes (intervalo QTc ≤330 ms).
-
Distancia desde el punto J al ápice de T < 120 ms (muy corta por la ausencia o casi ausencia de segmento ST).
-
Presencia del signo minus-plus de la onda T, es decir, onda T bifásica con la primera porción negativa y la final positiva10 (Figura 4).
-
Alta prevalencia del patrón de repolarización precoz (PRP) asociado20.
-
Eventual depresión del segmento PR.
-
Prolongación del intervalo pico de T/fin de T (TpTe: T peak to T end
-
Eventual onda U prominente.
-
Respuesta a la prueba de esfuerzo con retardada reducción del intervalo QTc durante el aumento en la FC.
-
Períodos refractarios auriculares y ventriculares muy cortos y tendencia a FA y FV inducibles en estudios electrofisiológicos (EEF).
-
Autopsia negativa. El diagnóstico de certeza solo posible con la autopsia molecular21.
-
En pacientes sintomáticos el cardiodesfibrilador implantable (CDI) es el tratamiento de elección.
-
Puede estar indicado quinidina, disopiramida y eventualmente en asociación con betabloqueantes.
Mutaciones en el canal de Ca2+ tipo L (LTCC: L-type calcium channel) responsables por las variantes SQTC4, SQTC5 y SQTC6
Las mutaciones que afectan el canal lento de entrada de Ca2+ en fase 2 pueden ocasionar los fenotipos del síndrome de Brugada (SBr), síndrome de Timothy (LQT8), síndrome de hipertermia maligna22, SQTC23 y los síndromes de onda J o de repolarización precoz (SRP; en inglés: early repolarization syndrome)24 frecuentemente asociados a arritmias cardíacas hereditarias, lo que sugiere que el mapeo genético, screening, de los genes que controlan los canales lentos de Ca2+ puede ser una valiosa herramienta diagnóstica en la identificación de individuos en riesgo. Una mutación en el gen CACNA2D1 puede ocasionar el SBr y las mutaciones en los genes CACNA1C, CACNB2 y CACNA2D1 pueden aumentar la susceptibilidad al SRP.
Las mutaciones con pérdida de función en el canal lento de Ca2+ (ICa) tipo L en los genes CACNB2b o CACNA1C pueden resultar en un fenotipo mixto de SBr asociado a un intervalo QT más corto de lo normal (overlapping). Las mutaciones del canal de Ca2+ lento que originan las posibles variantes SQTC4 y SQTC5 producen un fenotipo combinado de SQTC/SBr. El fenotipo del SBr y una historia familiar de MCS se asociaron con QTc≤360 ms2. En estos casos, mutaciones en los genes CACNB2b (S481L) y CACNA1C (A39V y G490R), que codifican las subunidades α1- o β2b- del canal de Ca2+ cardíaco tipo L fueron identificados.
Mutaciones en el gen CACNA1C se han asociado con el síndrome de Timothy (LQT8), una entidad caracterizada por disfunción multiorgánica, como cardiopatía congénita, arritmias letales, membranas interdigitales, deficiencia inmunológica, hipoglucemia intermitente, anormalidades cognitivas y autismo25.
La Figura 5 muestra el ECG en la derivación precordial intermedia V3 de la variante SQTC5, destacando la importancia de la medición de los intervalos Q-aT (inicio del QRS al ápice de T) y Q-oT (inicio del QRS al comienzo de T), los cuales están siempre muy cortos, imitando el ECG de la hipercalcemia severa.
Para el diagnóstico de SQTC, especialmente aquellos con intervalos QT limítrofes, las causas adquiridas de intervalo QT corto deben ser excluidas. La Tabla 2 relaciona las causas adquiridas de intervalo QT/QTc cortos.
Intervalos QT cortos en otras canalopatías
Sabemos que otras canalopatías pueden presentar intervalos QT/QTc cortos. Entre ellas destacamos:
• Fibrilación ventricular idiopática (FVI): valores QTc “cortos” se observan comúnmente en hombres con FVI. Sin embargo, valores de QTc “cortos” no son raros entre adultos sanos, especialmente durante la bradicardia. Viskin et al. demostraron que hombres portadores de FVI tienen intervalos QTc más cortos que los controles normales (371±22 ms)26. Estos autores sugirieron valores de corte de 360 ms para hombres y 370 ms para mujeres para considerar un intervalo QT como corto.
• Síndrome de Brugada tipo 3 (CACNA1C/Brugada syndrome 3): esta variante del SBr es causada por mutación heterocigota en la subunidad α-1C del gen CACNA1C que codifica el canal de Ca2+ lento o tipo L, localizado en el cromosoma 12p13. Fue identificada por Antzelevitch et al.2 Estos autores informaron que los probandos con SBr también presentaban intervalos QT más cortos.
• Síndrome de repolarización precoz: hay una alta prevalencia de repolarización precoz en pacientes portadores de SQTC. Además, el PRP puede ser útil en la identificación de eventos cardíacos20. Debemos estar muy atentos a aquellos pacientes con PRP y elevaciones del punto J>2,0 mm de convexidad superior (el PRP benigno es siempre de concavidad superior), especialmente aquellos con arritmias o historia familiar de MCS no explicadas.
Intervalo desde el punto J al pico de T
El intervalo punto J-pico de T es la distancia desde el punto J hasta el pico de T. Valores < 120 ms son valiosos para el diagnóstico del SQTC congénito27.
La Tabla 3 muestra el sistema de puntaje (score) para el diagnóstico del SQTC propuestos por Gollob27.
Prolongación del intervalo pico de T/fin de T (Tpe)
El posible sustrato para el desarrollo de taquiarritmias ventriculares puede ser una dispersión transmural significativa de la repolarización por acortamiento heterogéneo de la duración del PA. Normalmente el intervalo pico de T/fin de T es de 94 ms en hombres y 92 ms en mujeres cuando se mide en V5. En el SQTC, este parámetro está prolongado: >92 ms en mujeres y >94 ms en hombres con la medición en V5. El intervalo TpTe debe medirse en las derivaciones precordiales28. La prolongación a valores ≥120 ms se asocia a un mayor número de eventos en portadores de canalopatías.
Índice T(p-e)/QT
Es un índice electrocardiográfico de arritmogénesis, de enfermedades congénitas y adquiridas de canales iónicos que resultan en arritmias ventriculares. En los individuos sanos el índice T(p-e)/QT presenta un valor promedio de aproximadamente 0,21 en las precordiales y permanece relativamente constante entre las FC de 60 a 100 lpm.
El índice T(p-e)/QT es significativamente mayor en pacientes en riesgo de evento arrítmico como aquellos con SQTL, síndrome de Brugada, SQTC y también en pacientes con cardiopatía orgánica como infarto agudo de miocardio29 e hipertrofia del VI30 y apnea obstructiva del sueño (AOS)31.
Posible onda U prominente
La onda U es la última, inconstante y menor deflexión del ECG que se registra inmediatamente luego de la onda T y antes de la onda P del ciclo siguiente, de igual polaridad a la T precedente, es decir positiva cuando T lo es. El voltaje de U es siempre menor que el 50% de la amplitud de la T precedente y generalmente entre el 5% y el 25% de ella. Generalmente no supera 1 mm, siendo en promedio de 0,33 mm. Si alcanza 1,5 mm o más, es considerado alto; sin embargo, puede haber ondas U normales de hasta 2 mm (0,2 mV) en II y de V2 a V4.
Presencia de arritmias en el SQTC
Arritmias supraventriculares: se presentan episodios de FA paroxística, FA y respuesta ventricular lenta. Tanto los pacientes con síndromes hereditarios de QT prolongado como corto, que representan los extremos del intervalo QT, muestran una aparente alta prevalencia de FA.
Tratamiento
El SQTC conlleva un alto riesgo de MCS por eventos arrítmicos fatales y una alta penetrancia en las familias afectadas. Por lo tanto, la terapia con CDI es el apoyo principal para estos pacientes. Si bien esto es evidente en los sintomáticos, las dudas surgen en los asintomáticos, especialmente en ausencia de antecedentes familiares. Hasta el momento, no hay suficiente evidencia para la estratificación del riesgo evidente dado el bajo número de casos documentados y su diagnóstico relativamente reciente. En general se acepta que las manifestaciones clínicas, historia familiar y un EEF positivo o prueba genética pueden apoyar la implantación de un CDI. Sin embargo, un resultado negativo no excluye el diagnóstico o la posibilidad de futuros eventos arrítmicos.
A pesar de que el CDI es el único tratamiento eficaz, también tiene problemas específicos en estos pacientes. Por un lado, algunos informes mostraron un aumento del riesgo de descargas inapropiadas por taquicardia sinusal, FA y, sobre todo, sobredetección de las ondas T, que a menudo son altas y estrechas. En el estudio publicado por Schimpf et al.32, 3 de 5 pacientes recibieron choques inapropiados debido a la sobredetección de la onda T. La razón era una reducción posoperatoria de la amplitud de onda R y el aumento de la señal de la onda T. Por lo tanto, la adaptación de la programación estándar para prevenir la sobredetección de la onda T debe ser considerada después de la implantación y durante el seguimiento, aunque garantizando siempre una detección adecuada de las arritmias ventriculares. Por otra parte, la implantación de CDI en los niños aumenta las dificultades técnicas y complicaciones y no es factible en los más jóvenes.
Aunque el implante de un CDI es la terapia de primera línea, el tratamiento farmacológico puede estar indicado en algunos casos:
-
Como alternativa a la implantación de un CDI en los niños pequeños.
-
Pacientes con contraindicaciones o para una disminución del implante de un CDI.
-
Como terapia adjunta para evitar las descargas excesivas.
-
Prevención de los episodios sintomáticos de FA.
Los fármacos deben usarse con precaución ya que la eficacia a largo plazo del tratamiento farmacológico en la prevención de eventos arrítmicos graves no está bien establecida. La quinidina es la terapia farmacológica más eficaz. Esta droga bloquea varios canales de potasio (IKr, Ito, IKATP e IK1) y las corrientes de sodio y de calcio hacia el interior.
En la variante SQTC1 la quinidina ocasiona una marcada prolongación del segmento ST, onda T, intervalo QT y de los períodos refractarios efectivos ventriculares, así como restaura la adaptación de la dependencia de la FC del intervalo QT, ocasiona disminución de la dispersión de la repolarización (Tp/Te) y previene de la inducción de FV. Sin embargo, se desconocen las consecuencias clínicas de estos efectos electrofisiológicos.
La disopiramida prolonga el intervalo QT y el período refractario efectivo ventricular en la variante SQTC1, de modo que es una alternativa a la quinidina33 en los países donde esta droga no se comercializa.
Las mutaciones N588K-KCNH2 y V307L-KCNQ1 que ocasionan una ganancia de función de IKr y IKs, causantes de las variantes SQTC1 y SQTC2, se benefician con el tratamiento farmacológico, asociando los bloqueantes de los canales de K+ (como quinidina) a los betabloqueantes34.
La propafenona ha demostrado ser eficaz en la prevención de paroxismos frecuentes de FA sin recurrencia de la arritmia durante más de dos años, y sin ejercer ningún efecto sobre el intervalo QT35.
(Esta revisión cuenta con material adicional, disponible en nuestrio sitio web: http://www.conarec.org/).
Conclusión
A lo largo de esta revisión presentamos las principales características genéticas, clínicas, electrocardiográficas y electrofisiológicas de la más reciente y rara canalopatía cardíaca sin cardiopatía estructural. Describimos las formas que afectan los canales de K+ y Ca2+ en sus características especiales. Abordamos los valores de QT, QTc, la elevada tendencia a la aparición de FA paroxística, síncope y MCS. Mostramos el sistema de puntaje de Gollob, que ayuda en el diagnóstico. Destacamos los valores extremadamente cortos del intervalo QT/QTc, la falta de adaptación del QTc a la súbita modificación de la FC, la disminución del valor del intervalo entre el punto J y ápice de T, la elevada prevalencia del PRP, el aspecto peculiar de las ondas T altas y de base estrecha, el signo minus-plus de la onda T, y la dispersión transmural de la repolarización ventricular manifestada por prolongación del intervalo Tp-Te y la eventual presencia de onda U prominente. Finalmente, mostramos que el diagnóstico de certeza post mortem exige las nuevas técnicas de autopsia molecular y que el tratamiento de elección en los sintomáticos es el implante de CDI asociado en casos específicos a quinidina.
-
Schimpf R, Kuschyk J, Veltmann C, Borggrefe M, Wolpert C. Primary electrical heart disease in adulthood-electrophysiological findings and therapy. Herzschrittmacherther Elektrophysiol. 2005;16(4):250-9.
-
Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115(4):442-9.
-
Giustetto C, Schimpf R, Mazzanti A, Scrocco C, Maury P, Anttonen O, et al. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol. 2011; 58(6):587–95.
-
Spears DA, Gollob MH. Genetics of inherited primary arrhythmia disorders. Appl Clin Genet. 2015;8:215-33.
-
Bjerregaard P. Proposed diagnostic criteria for short QT syndrome are badly founded. J Am Coll Cardiol. 2011; 58(5):549-50; author reply 550-1.
-
Shimizu W. Atrial fibrillation and genetic abnormalities. Nihon Rinsho. 2013;71(1):161-6.
-
Kobza R, Roos M, Niggli B, Abächerli R, Lupi GA, Frey F, et al. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm. 2009;6(5):652–7.
-
Chinushi M, Sato A, Izumi D, Furushima H. Nifekalant enlarged the transmural activation-recovery interval difference as well as the peak-to-end interval on surface ECG in a patient with short-QT syndrome. J Cardiovasc Electrophysiol. 2012;23(8):877-80.
-
Pérez Riera AR, Ferreira C, Dubner SJ, Schapachnik E, Soares JD, Francis J. Brief review of the recently described short QT syndrome and other cardiac channelopathies. Ann Noninvasive Electrocardiol. 2005;10(3):371-7.
-
Pérez Riera AR, Paixão-Almeida A, Barbosa-Barros R, Yanowitz FG, Baranchuk A, Dubner S, et al. Congenital short QT syndrome: landmarks of the newest arrhythmogenic cardiac channelopathy. Cardiol J. 2013;20(5):464-71.
-
Anttonen O, Junttila J, Giustetto C, Gaita F, Linna E, Karsikas M, et al. T-Wave morphology in short QT syndrome. Ann Noninvasive Electrocardiol. 2009;14(3):262-7.
-
Gaita F, Giustetto C, Bianchi F, Wolpert C, Schimpf R, Riccardi R, et al. Short QT syndrome: a familial cause of sudden death. Circulation 2003;108(8):965-70.
-
Gaita F, Giustetto C, Bianchi F, Schimpf R, Haissaguerre M, Calò L, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol. 2004;43(8):1494-9.
-
Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 2004;109(1):30-5.
-
Wilders R. Cardiac ion channelopathies and the sudden infant death syndrome. ISRN Cardiol. 2012;2012:846171.
-
Bellocq C, van Ginneken AC, Bezzina CR, Alders M, Escande D, Mannens MM, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004;109(20):2394-7.
-
Maltret A, Wiener-Vacher S, Denis C, Extramiana F, Morisseau-Durand MP, Fressart V, et al. Type 2 short QT syndrome and vestibular dysfunction: mirror of the Jervell and Lange-Nielsen syndrome? Int J Cardiol. 2014;171(2):291-3.
-
Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005;96(7):800-7.
-
Hattori T, Makiyama T, Akao M, Ehara E, Ohno S, Iguchi M, et al. A novel gain-of-function KCNJ2 mutation associated with short-QT syndrome impairs inward rectification of Kir2.1 currents. Cardiovasc Res. 2012;93(4):666-73.
-
Watanabe H, Makiyama T, Koyama T, Kannankeril PJ, Seto S, Okamura K, et al. High prevalence of early repolarization in short QT syndrome. Heart Rhythm. 2010;7(5):647-52.
-
Farrugia A, Keyser C, Hollard C, Raul JS, Muller J, Ludes B. Targeted next generation sequencing application in cardiac channelopathies: Analysis of a cohort of autopsy-negative sudden unexplained deaths. Forensic Sci Int. 2015;254:5-11.
-
Robinson RL, Curran JL, Ellis FR, Halsall PJ, Hall WJ, Hopkins PM, et al. Multiple interacting gene products may influence susceptibility to malignant hyperthermia. Ann Hum Genet.2000;64(Pt 4):307–20.
-
Templin C, Ghadri JR, Rougier JS, Baumer A, Kaplan V, Albesa M, et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur Heart J. 2011;32(9):1077-88.
-
Burashnikov E, Pfeiffer R, Barajas-Martinez H, Delpón E, Hu D, Desai M, et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm. 2010;7(12):1872-82.
-
Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119(1):19–31.
-
Viskin S, Zeltser D, Ish-Shalom M, Katz A, Glikson M, Justo D, et al. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fibrillation and healthy controls. Heart Rhythm. 2004;1(5):587-91.
-
Gollob MH, Redpath CJ, Roberts JD. The Short QT syndrome Proposed Diagnostic Criteria. J Am CollCardiol. 2011;57(7):802-12.
-
Bieganowska K, Sawicka-Parobczyk M, Bieganowski M, Piskorski J. T peak -tend interval in 12-lead electrocardiogram of healthy children and adolescents t peak -tend interval in childhood. Ann Noninvasive Electrocardiol. 2013;18(4):344-51.
-
Gupta P, Patel C, Patel H, Narayanaswamy S, Malhotra B, Green JT, et al. T(p-e)/QT ratio as an index of arrhythmogenesis. J Electrocardiol. 2008;41(6):567-74.
-
Zhao Z, Yuan Z, Ji Y, Wu Y, Qi Y. Left ventricular hypertrophy amplifies the QT, and Tp-e intervals and the Tp-e/ QT ratio of left chest ECG. J Biomed Res. 2010;24(1):69-72.
-
Kilicaslan F, Tokatli A, Ozdag F, Uzun M, Uz O, Isilak Z, et al. Tp-e interval, Tp-e/QT ratio, and Tp-e/QTc ratio are prolonged in patients with moderate and severe obstructive sleep apnea. Pacing Clin Electrophysiol. 2012;35(8):966-72.
-
Schimpf R, Antzelevitch C, Haghi D, Giustetto C, Pizzuti A, Gaita F, et al. Electromechanical coupling in patients with the short QT syndrome: further insights into the mechanoelectrical hypothesis of the U wave. Heart Rhythm. 2008;5(2):241-5.
-
Mizobuchi M, Enjoji Y, Yamamoto R, Ono T, Funatsu A, Kambayashi D, et al. Nifekalant and disopyramide in a patient with short QT syndrome: evaluation of pharmacological effects and electrophysiological properties. Pacing Clin Electrophysiol. 2008;31(9):1229-32.
-
Bodi I, Franke G, Pantulu ND, Wu K, Perez-Feliz S, Bode C, et al. Differential effects of the β-adrenoceptor blockers carvedilol and metoprolol on SQT1- and SQT2-mutant channels. J Cardiovasc Electrophysiol. 2013;24(10):1163-71.
-
Bjerregaard P, Nallapaneni H, Gussak I. Short QT interval in clinical practice. J Electrocardiol. 2010;43(5):390-5.
Bibliografía del Material Suplementario
-
Gussak I, Brugada P, Brugada J, Wright RS, Kopecky SL, Chaitman BR, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology 2000;94(2):99-102.
-
Hong K, Bjerregaard P, Gussak I, Brugada R. Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J. Cardiovasc Electrophysiol 2005;16(4):394-6.
-
Gaita F, Giustetto C, Bianchi F, Wolpert C, Schimpf R, Riccardi R, et al. Short QT syndrome: a familial cause of sudden death. Circulation 2003;108(8):965-70.
-
Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 Circ Res 2005;96(7):800-7.
-
Templin C, Ghadri JR, Rougier JS, Baumer A, Kaplan V, Albesa M, et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur Heart J 2011;32(9):1077-88.
-
Robinson RL, Curran JL, Ellis FR, Halsall PJ, Hall WJ, Hopkins PM, et al. Multiple interacting gene products may influence susceptibility to malignant hyperthermia. Ann Hum Genet 2000;64(Pt 4):307-20.
-
Burashnikov E, Pfeiffer R, Barajas-Martinez H, Delpón E, Hu D, Desai M, et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 2010;7(12):1872-82.
-
Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007;115(4):442-9.
-
Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007;115(4):442-9.
-
Watanabe H, Makiyama T, Koyama T, Kannankeril PJ, Seto S, Okamura K, et al. High prevalence of early repolarization in short QT syndrome. Heart Rhythm 2010;7(5):647-52.
-
Pérez MV, Friday K, Froelicher V. Semantic confusion: the case of early repolarization and the J point. Am J Med 2012;125(9):843-4.
-
Davey P, Bateman J. Heart rate and catecholamine contribution to QT interval shortening on exercise. Clin Cardiol 1999;22(8):513-8.
-
Gollob MH, Redpath CJ, Roberts JD. The Short QT syndrome proposed diagnostic criteria. J Am Coll Cardiol 2011;57(7):802-12.
-
Bieganowska K, Sawicka-Parobczyk M, Bieganowski M, Piskorski J. T peak -tend interval in 12-lead electrocardiogram of healthy children and adolescents t peak -tend interval in childhood. Ann Noninvasive Electrocardiol 2013;18(4):344-51.
-
Anttonen O, Vaananen H, Junttila J, Huikuri HV, Viitasalo M. Electrocardiographic transmural dispersion of repolarization in patients with inherited short QT syndrome. Ann Noninvasive Electrocardiol 2008;13(3):295-300.
-
Chinushi M, Sato A, Izumi D, Furushima H. Nifekalant enlarged the transmural activation-recovery interval difference as well as the peak-to-end interval on surface ECG in a patient with short-QT syndrome. J Cardiovasc Electrophysiol 2012;23(8):877-80.
-
Gupta P, Patel C, Patel H, Narayanaswamy S, Malhotra B, Green JT, et al. T(p-e)/QT ratio as an index of arrhythmogenesis. J Electrocardiol 2008;41(6):567-74.
-
Zhao Z, Yuan Z, Ji Y, Wu Y, Qi Y. Left ventricular hypertrophy amplifies the QT, and Tp-e intervals and the Tp-e/QT ratio of left chest ECG. J Biomed Res 2010;24(1):69-72.
-
Kilicaslan F, Tokatli A, Ozdag F, Uzun M, Uz O, Isilak Z, et al. Tp-e interval, Tp-e/QT ratio, and Tp-e/QTc ratio are prolonged in patients with moderate and severe obstructive sleep apnea. Pacing Clin Electrophysiol 2012;35(8):966-72.
-
Zhao X, Xie Z, Chu Y, Yang L, Xu W, Yang X, et al. Association between Tpe/QT ratio and prognosis in patients undergoing primary percutaneous coronary intervention for ST-segment elevation myocardial infarction. Clin Cardiol 2012;35(9):559-64.
-
Bellocq C, van Ginneken AC, Bezzina CR, Alders M, Escande D, Mannens MM, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 2004;109(20):2394-7.
-
Hong K, Piper DR, Diaz-Valdecantos A, Brugada J, Oliva A, Burashnikov E, et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc Res 2005;68(3):433-40.
-
Antzelevitch C, Nesterenko VV, Yan GX. Role of M cells in acquired long QT syndrome, U waves, and torsade de pointes. J Electrocardiol 1995;28 Suppl:131-8.
-
Schimpf R, Antzelevitch C, Haghi D, Giustetto C, Pizzuti A, Gaita F, et al. Electromechanical coupling in patients with the short QT syndrome: further insights into the mechanoelectrical hypothesis of the U wave. Heart Rhythm 2008;5(2):241-5.
-
Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114(20):2104-12.













Variantes del síndrome de QT corto congénito
La Tabla 1 - Material suplementario (MS) muestra las variantes identificadas del síndrome de QT corto (SQTC) congénito con sus respectivos valores medios de los intervalos QTc, símbolo genético, autor y año del descubrimiento. Las tres primeras variantes que afectan los canales de potasio (K+) son consideradas indiscutiblemente integrantes del SQTC. Las variantes 4 y 5, que afectan el canal de calcio (Ca2+) lento, en realidad, son síndromes de superposición fenotípica, overlapping, (síndrome de Brugada [SBr] + SQTC); y la variante 6, también afectadora del canal de Ca+2, tiene un intervalo QTc no tan corto de 329 ms.
Nomenclatura OMIM (Online Mendelian Inheritance in Man)
El proyecto OMIM es una base de datos que cataloga todas las enfermedades humanas con componente genético. Cuando es posible, estas enfermedades se relacionan con sus genes. A cada enfermedad y al gen se le asigna un número de seis dígitos. El primer número refleja el tipo de herencia. La Tabla 2 y 3 - MS muestra el número OMIM, nombre del gen y su locus.
Síndrome de QT corto congénito tipo 1 o fenotipo SQTC1 (IKr); casos reportados
Gussak et al.1 informaron sobre dos hermanos (hermano y hermana adolescentes) y su madre que presentaban un intervalo QT/QTc idiopático, persistentemente muy corto (280, 272 y 260 ms, respectivamente). La hermana de 17 años presentaba repetidos episodios de fibrilación auricular (FA) paroxística que fueron revertidos con cardioversión eléctrica.
Hong et al.2 informaron que en la familia estudiada originalmente por Gussak et al. el abuelo materno ya fallecido también presentaba intervalo QT corto y FA crónica, y la estimulación eléctrica programada en la madre y los dos hermanos reveló un período refractario auricular y ventricular muy cortos e inducibilidad de FA y fibrilación ventricular (FV). Los tres recibieron cardiodesfibrilador implantable (CDI) y tratamiento con propafenona, que los mantuvo libres de eventos de FA paroxística.
Gaita et al.3 describieron 2 árboles genealógicos de 5 generaciones de una familia con fuerte historia de muerte súbita cardiaca (MCS) e intervalos QT muy cortos, idiopáticos, sin cardiopatía estructural. Las manifestaciones incluyeron palpitaciones, síncope y MCS por paro cardíaco. Se registraron MCS en ambos sexos, en 4 generaciones, con transmisión de padre a hijo en ambas familias, lo que sugiere una herencia autosómica dominante.
Seis integrantes de las familias se sometieron a una evaluación extensa, y todos mostraron QTc ≤ 280 ms, además de períodos refractarios auriculares y ventriculares muy cortos con vulnerabilidad ventricular aumentada para FA y FV en 3 de 4 integrantes.
Síndrome de QT corto congénito fenotipo tipo 3 o SQTC3 (IK1); casos reportados
Priori et al.4 identificaron una mutación missense en el gen KCNJ2. La mutación no estuvo presente en la madre no afectada o en los abuelos paternos, indicando que puede haber ocurrido una mutación de novo. Los miembros afectados de una única familia tenían una sustitución G514A en el gen KCNJ2 consistente en la sustitución del ácido aspártico en la posición 172 (D172N). Esta es considerada la tercera variante del SQTC (SQTC3). Estas mutaciones se observaron en una niña asintomática de 5 años que presentaba un intervalo QTc de 315 ms y ondas T de base estrecha y amplias, y en el padre de 35 años con un intervalo QTc corto (320 ms) además de historia de episodios presincopales y palpitaciones desde la adolescencia. Los electrocardiogramas (ECG) de la niña (probando) y su padre se caracterizaban por ondas T asimétricas con rampa ascendente bastante normal y rampa terminal rápidamente descendente. La madre y abuelos paternos tenían ECG normales e informaron no tener historia familiar de MCS.
Mutaciones en el canal de Ca2+ tipo L (LTCC: L-type calcium channel) ocasionadoras de las variantes SQTC4, SQTC5 y SQTC6
La Tabla 4 - MS muestra las variantes 4, 5 y 6 del SQTC por mutaciones que afectan el canal lento de Ca2+, su símbolo genético y efecto sobre el canal, así como el autor de la identificación.
La Figura 1 - MS muestra las características electrocardiográficas de la hipercalcemia grave, la cual puede ser indistinguible electrocardiográficamente con la variante a SQTC5, es decir, con intervalos Q-aT, Q-oT y QTc muy cortos. La hipercalcemia es una de las causas adquiridas de intervalo QT corto.
Tabla 4 - MS. Variantes 4, 5 y 6 del SQTC por mutaciones que afectan el canal lento de Ca2+, símbolo genético, efecto sobre el canal, y autor de la identificación.
SQTC4: símbolo genético: CACNA1C. Antzelevitch C et al. Circulation 2007;115:442.
SQTC5: símbolo genético: CACNA1C (ICa); Nombre del gen: canal de calcio dependiente de voltaje, tipo L, subunidad alpha 1C. Antzelevitch et al. Circulation 2007;115:442.
SQTC6: símbolo genético: CACNA2D1 (ICa): Nombre del gen: canal de calcio dependiente de voltaje, subunidad α2/δ1 5. Además, la mutación en CACNA2D1 se ha asociado con susceptibilidad a hipertermia maligna6 y síndrome de repolarización precoz7.
Intervalos QT cortos en otras canalopatías
-
Síndrome de Brugada tipo 3 - CACNA1C/Brugada syndrome 3: esta variante del SBr es causada por mutación heterocigota en la subunidad α-1C del gen CACNA1C que codifica el canal de Ca2+ lento o tipo L, localizado en el cromosoma 12p13. Fue identificada por Antzelevitch el al.9 Estos autores informaron que los probandos con SBr también presentaban intervalos QT más cortos. Estudiaron un hombre de 41 años de ascendencia turca, con FA y un QTc de 346 ms. La administración de ajmalina resultó en el patrón tipo 1 en las precordiales derechas y la aparición de una ráfaga de taquicardia ventricular (TV) monomórfica. Este paciente tenía un hermano que murió de paro cardíaco a los 45 años y dos hijas con intervalos QTc cortos (360 y 373 ms, respectivamente). El otro probando era un hombre de 44 años de ascendencia europea, con patrón de Brugada tipo 2 en V2, onda J prominente en III y QTc de 360 ms. Este era portador de distrofia muscular fascio-escápulo-humeral. La madre tuvo 2 episodios sincopales a los 48 años, que resultaron en MCS; y el padre, 2 hermanos y 3 hijos se negaron a ser examinados, pero aparentemente no exhibían el fenotipo de Brugada.
-
Síndrome de repolarización precoz: en el concepto antiguo clásico, el patrón electrocardiográfico de repolarización precoz (PRP) era considerado presente ante la elevación del punto J y segmento ST ≥ 1,0 mm (0,1 mV) en por lo menos dos derivaciones contiguas, sea de la pared inferior o lateral, y asociada a un empastamiento o muesca en la parte final de la rampa descendente de la R del complejo QRS. Hay una alta prevalencia de repolarización precoz en pacientes portadores de SQTC. Además, el PRP puede ser útil en la identificación de eventos cardíacos10. Debemos estar muy atentos a aquellos pacientes con PRP y elevaciones del punto J > 2,0 mm, y de convexidad superior (el PRP benigno es siempre de concavidad superior) especialmente aquellos con arritmias no explicadas o historia familiar de MCS no explicada. Los pacientes con PRP presentan más probabilidades de ser hombres, de haber experimentado síntomas durante el sueño y de presentar intervalo QTc más corto que aquellos sin PRP. En la actualidad no es necesaria la presencia de elevación del ST para caracterizar el diagnóstico de PRP11. La Figura 2 - MS compara los conceptos clásicos (A y B) y actuales (C y D) del PRP. Destaquemos que en el SQTC la prevalencia del PRP es elevada. La Figura 3 - MS muestra un ECG de un paciente portador del SQTC asociado a PRP.
Intervalo QT
El intervalo QT es un índice de la repolarización ventricular parcialmente dependiente de la frecuencia cardíaca (FC); en otras palabras, se acorta con el ejercicio y se prolonga con la bradicardia. Una parte de esta reducción se debe al aumento de la FC, y otra parte, a otros efectos producidos durante el ejercicio, probablemente efectos neuroendocrinos. En corazones normales, dos tercios del acortamiento del QT son ocasionados por el ejercicio y un tercio por otros factores. Los cambios en los niveles de catecolaminas en plasma con el ejercicio no se relacionan estrechamente con los cambios en la duración del QT durante el ejercicio12. En comparación con la población normal, los pacientes portadores del SQTC tienen menos variación del intervalo QTc en relación con los cambios en la FC. Esto da lugar a una interpretación errónea del intervalo QTc en presencia de una FC más rápida y el diagnóstico de falsos negativos.
Intervalo desde el punto J al pico de T corto
El intervalo punto J-pico de T es la distancia desde el punto J hasta el pico de T. Valores < 120 ms son valiosos para el diagnóstico del SQTC congénito13 (Figura 4 - MS).
Prolongación del intervalo pico de T/fin de T (Tpe)
El posible sustrato para el desarrollo de taquiarritmias ventriculares puede ser una dispersión transmural significativa de la repolarización por acortamiento heterogéneo de la duración del potencial de acción (DPA). Normalmente el intervalo pico de T/fin de T es de 94 ms en hombres y 92 en mujeres cuando se mide en V5. En el SQTC este parámetro está prolongado: > 92 ms en mujeres y > 94 ms en hombres con la medición en V5. En pacientes con SQTC1 las ondas T en las precordiales con frecuencia parecen altas, estrechas y simétricas, con un intervalo pico de T/fin de T prolongado3. La Figura 5 - MS muestra la correlación del intervalo pico de T y fin de T en el ECG de superficie con los potenciales de acción en el espesor de la pared ventricular y su valor normal.
En niños y adolescentes sanos, los intervalos TpTe varían entre las derivaciones individuales del ECG, siendo mayor en V3. El intervalo TpTe es más prolongado en niños y se prolonga a medida que la FC se desacelera. La dispersión TpTe varía entre 6 y 80 ms (promedio 38,6 ms ± 14,6 ms, mediana 40 ms) sin diferencias de género y valores mayores en personas de más edad. Los índices TpTe/QT y TpTe/JT son mayores en niños. El intervalo TpTe debe medirse en las derivaciones precordiales14. En adultos el valor normal del intervalo Tpe es 94 ms en hombres y 92 en mujeres cuando se mide en V5. La prolongación a valores ≥ 120 ms se asocia a un mayor número de eventos en portadores de canalopatías. Tpe puede corresponder a dispersión transmural de la repolarización ventricular (DTR) y, en consecuencia, la amplificación de este intervalo se asocia a arritmias ventriculares malignas.
La amplificación de la (DTR) es secundaria a prolongación preferencial de la DPA de las células M, mientras que, en el SBr, se piensa que se debe a acortamiento selectivo de la DPA del epicardio del VD. Estos parámetros indican DTR aumentada15. Sin embargo, también se ha informado sobre ondas T asimétricas con una rama ascendente menos aguda, seguidas de rama descendente rápida.
En el SQTC el acortamiento preferencial de la DPA del endocardio o el epicardio parecen ser responsables de la amplificación de la DTR.
En la taquicardia ventricular polimórfica catecolaminérgica, la inversión en la dirección de la activación de la pared ventricular es responsable del aumento en la DTR.
Así, los SQTL, SQTC, SBr y de la taquicardia ventricular polimórfica catecolaminérgica son patologías con diferentes fenotipos y etiologías; sin embargo, comparten una vía final común en su predisposición a la MCS.
La administración intravenosa de nifekalant prolonga un período refractario efectivo en múltiples sitios ventriculares, así como el intervalo QT/QTc (de 260/300 a 364/419 ms) en el ECG de superficie en el SQTC congénito. El nifekalant también aumentó la dispersión transmural del intervalo de recuperación de la activación, que se midió por la diferencia entre los intervalos de recuperación de activación endocárdica más prolongados y los más cortos durante estimulación auricular de 90 latidos por minuto (lpm), de 73 a 103-105 ms. Estos valores correspondieron a los intervalos entre el pico y el final de la onda T en el ECG de superficie. La prolongación de QT inducida por nifekalant en el ECG de superficie puede no indicar la atenuación del potencial arritmogénico en el corazón de pacientes con SQTC16.
Índice T(p-e)/QT
Es un índice electrocardiográfico de arritmogénesis, de enfermedades congénitas y adquiridas de canales iónicos que resultan en arritmias ventriculares. En los individuos sanos el índice T(p-e)/QT presenta un valor promedio de aproximadamente 0,21 en las precordiales y permanece relativamente constante entre las FC de 60 a 100 lpm.
El índice T(p-e)/QT es significativamente mayor en pacientes en riesgo de evento arrítmico como aquellos con SQTL, síndrome de Brugada, SQTC y también en pacientes con cardiopatía orgánica como infarto agudo de miocardio17 e hipertrofia del VI18 y apnea obstructiva del sueño (AOS)19.
Un índice Tp-e/QT ≥ 0,29 en pacientes que se someten a intervención coronaria percutánea primaria (ACTP) para el infarto agudo de miocardio con elevación del segmento ST (IAMEST) puede servir como un predictor pronóstico de resultados adversos luego de tratamiento exitoso con ACTP20.
La reentrada funcional es el mecanismo subyacente de la arritmogénesis asociada con un índice T(p-e)/QT aumentado.
Diferencias de morfologías de las ondas T en las variantes de SQTC que afectan los canales de K+
En el SQTC1, las ondas T en las derivaciones precordiales con frecuencia son altas, estrechas y simétricas, con un intervalo pico de T-fin de T corto3. La Figura 6 - MS muestra las características de la onda T en la variante SQTC1.
En el SQTC2 las ondas T parecen ser simétricas, pero no tan altas y estrechas21,22.
En el SQTC3 las ondas T son asimétricas con una rampa ascendente bastante normal y una rampa terminal descendente rápida4.
Posible onda U prominente
La onda U es la última, inconstante y menor deflexión del ECG que se registra inmediatamente luego de la onda T y antes de la onda P del ciclo siguiente, de igual polaridad a la T precedente, es decir positiva cuando T lo es. El voltaje de U es siempre menor que el 50% de la amplitud de la T precedente y generalmente entre el 5 y el 25% de ella. Generalmente no supera 1 mm, siendo en promedio de 0,33 mm. Si alcanza 1,5 mm o más, es considerado alto; sin embargo, puede haber ondas U normales de hasta 2 mm (0,2 mV) en II y de V2 a V4. La onda U se ubica inmediatamente luego de la onda T durante la fase protodiastólica del ciclo cardíaco (fase diastólica isovolumétrica y de llenado rápido) concomitante al segundo ruido y con la fase 4 del PA; frecuentemente ausente, ocasionalmente difícil de distinguir de la onda T precedente; se observa mejor durante bradicardia y a veces se relaciona con torsades de pointes (TdP). Además, la inscripción de la onda U en los pacientes con SQTC coincidió con el cierre de la válvula aórtica y con el período de relajación isovolumétrica, respaldando la hipótesis de que la onda U se relaciona con estiramiento mecánico. El intervalo desde el cierre de la válvula aórtica hasta el comienzo de la onda U fue 8±4 ms en pacientes con SQTC y 15±11 ms en sujetos de control.
-
Cuando la FC es ≤ 65 latidos por minuto (lpm) la onda U es visible en el 90% de los casos;
-
Cuando la FC está entre 80 y 95 lpm, la onda U es visible en el 65% de los casos;
-
Cuando la FC es > 96 lpm, la onda U es visible en el 25 % de los casos;
-
El SÂU en el plano horizontal se dirige hacia adelante y a la izquierda. De este modo la onda U se observa mejor en V3 (entre V2 y V4);
-
La onda U se observa mejor en las precordiales.
Causas de ondas U prominentes
-
Hipokalemia (recuerden la tríada de infradesnivel ST, ondas T de baja amplitud y ondas U prominentes). Ondas U anormalmente prominentes se observan característicamente en la hipokalemia grave.
-
Hipercalcemia.
-
Hipomagnesemia.
-
Hipotermia.
-
La bradicardia sinusal acentúa la onda U. La causa más común de ondas U prominentes es la bradicardia.
-
Inspiración forzada.
-
Posejercicio.
-
Antiarrítmicos clase 1A (quinidina, procainamida) y clase 3 (sotalol, amiodarona).
-
Fenotiazinas (tioridazina).
-
Presión intracraneal aumentada: en el contexto de hemorragia intracraneal. Enfermedad del SNC con intervalos QT prolongados (con frecuencia T y U se fusionan para formar una onda gigante de “fusión T-U”).
-
Hipertrofia del ventrículo izquierdo (HVI) (precordiales derechas con ondas S profundas).
-
Prolapso de la válvula mitral (algunos casos).
-
Hipertiroidismo, tirotoxicosis.
-
Exposición a digitálicos.
-
Epinefrina.
-
Síndrome de QT prolongado congénito.
-
Síndrome de QT prolongado adquirido23.
-
Síndrome de QT corto congénito24.
-
Bloqueo AV completo.
-
Miocardiopatía hipertrófica y otras miocardiopatías.
Presencia de arritmias en el SQTC
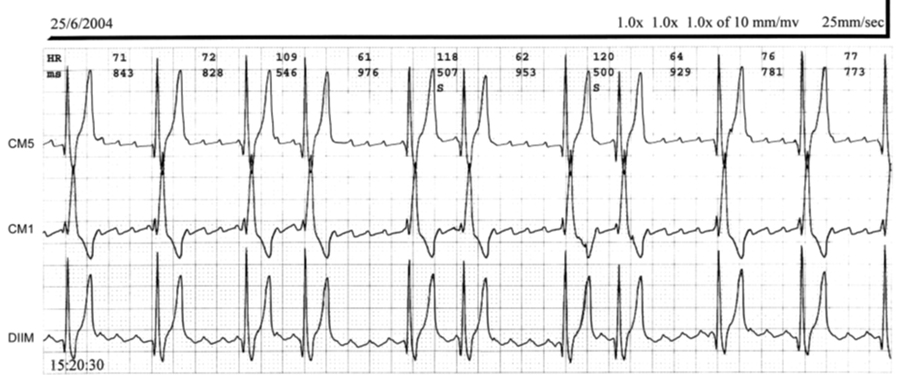
La Figura 7 - MS muestra un estudio de Holter de un paciente portador del SQTC durante un evento de FA paroxística.
Ejemplos de SQTC
Reporte de caso
Hombre caucásico, asintomático, de 44 años, natural de Victoria da Conquista, Bahía, Brasil.
Motivo de la consulta: derivado al cardiólogo para evaluar riesgo de biopsia de próstata bajo sedación. Asintomático.
Historia personal: aumento mínimo del antígeno prostático específico (PSA) en suero reciente. Chequeo. Examen rectal digital realizado por urólogo.
Historia familiar: fuerte historia de MCS en familiares de 1er grado: MCS en la madre a los 62 años, una hermana de 6 años y un hermano de 13 años. Dos hermanas asintomáticas de 36 y 41 años.
Examen físico: normal.
El ECG del paciente se muestra en la Figura 8 - MS.
Nombre: VTC; Sexo: Masc; Grupo étnico: caucásico; Edad: 44 años (de Bahía, Brasil, 12 de febrero de 1968); Peso: 84 kg; Altura: 1,79 m; Fecha: 19 de abril de 2012; Drogas en uso: ninguna. ECG del probando, caso índice o propósitus.
La Figura 9 - MS muestra el ECG de una hermana mayor del probando. Nombre: MTC; Sexo: Fem; Edad: 54 años; Fecha: 20 de marzo de 2014; Grupo étnico: caucásico.
La Figura 10 - MS muestra el ECG de un hijo del probando con un patrón altamente sugestivo de SQTC. Nombre: WTC; Sexo: Masc; Grupo étnico: caucásico; Edad: 23 años (de Bahía, Brasil, 21 de marzo de 1989); Peso: 68 kg; Altura: 1,72 m; Fecha: 24 de abril de 2012; Drogas en uso: ninguna.
Laboratório de Delineamento de Estudos e Escrita Científica na Faculdade de Medicina do ABC, Fundação do ABC. Santo André, São Paulo, Brasil.
Laboratório de Delineamento de Estudos e Escrita Científica na Faculdade de Medicina do ABC, Fundação do ABC. Santo André, São Paulo, Brasil. Visiting Scientist at Program in Molecular and Integrative Physiological Sciences (MIPS), Department of Environmental Health, Harvard T.H. Chan School of Public Health..
Chefe da unidade coronariana do Hospital de Messejana Dr. Carlos Alberto Studart Gomes. Fortaleza, Ceará, Brasil..
FESC - Unidade Medico-Cirúrgica De Conquista - Diretor da Divisão de Cardiologia e Eletrocardiografia - Sociedade Regional de Cardiologia do Sudoeste SBC/BA Brasil. Diretor da Comissão Cientifica..
Autor correspondencia
Laboratório de Delineamento de Estudos e Escrita CientÃfica na Faculdade de Medicina do ABC, Fundação do ABC. Santo André, São Paulo, Brasil.
Correo electrónico: Rua Sebastião Afonso 885 CEP 04417-100. Jardim Miriam São Paulo Brasil | Fone 55-11-56212390 | riera@uol.com.br
Para descargar el PDF del artículo
El síndrome del QT corto congénito: avances en los últimos años
![]() Haga click aquí
Haga click aquí
Para descargar el PDF de la revista completa
Revista del CONAREC, Volumen Año 2016 Num 135

Revista del CONAREC
Número 135 | Volumen
31 | Año 2016

Editorial
María Florencia González Amigo
Tratamiento fibrinolítico en el tr...
Mario César Spennato y cols.
El síndrome del QT corto congénit...
Andrés R Pérez-Riera y cols.
Antiagregación en implantación va...
Sol Pinasco
El índice de masa ventricular como...
Agustina Ginesi y cols.
Seguimiento a 8 años de aneurismas...
José Picco y cols.
Disección arterial coronaria espon...
Cecilia Andreani y cols.
Miocarditis aguda de etiología cha...
Pablo Pulenta y cols.
Una causa infrecuente de disnea
Sebastián García Zamora y cols.
Etiquetas
canalopatÃas, sÃndrome de QT corto, fibrilación auricular, muerte cardiaca súbita
Tags
channelopathies, short QT syndrome, atrial fibrillation, sudden cardiac death

El síndrome del QT corto congénito: avances en los últimos años
Autores
Andrés R Pérez-Riera, Luiz C De Abreu, Raimundo Barbosa-Barros, Adail Paixão-Almeida
Publicación
Revista del CONAREC
Editor
Consejo Argentino de Residentes de Cardiología
Fecha de publicación
2016-09-01
Registro de propiedad intelectual
© Consejo Argentino de Residentes de Cardiología






Reciba la revista gratis en su correo

Suscribase gratis a nuestra revista y recibala en su correo antes de su publicacion impresa.
