Artículo de Revisión
Síndrome de QTc largo
William Uribe, Jorge Enrique Velásquez, Mauricio Duque, Adrián Baranchuk
Revista del Consejo Argentino de Residentes de CardiologÃa 2016;(133): 0015-0020
El Síndrome de QT largo (SQTL) es una canalopatía responsable de la prolongación anormal de la repolarización ventricular con un comportamiento hereditario muy importante. Este artículo revisa de manera profunda los hallazgos electrocardiográficos y genéticos que permitirán al lector aumentar sus conocimientos con el fin de diagnosticar y clasificar de la mejor manera posible a su paciente con síndrome de QT largo. Sus manifestaciones clínicas van desde palpitaciones hasta episodios sincopales, los cuales pueden dar como resultado paro cardiaco y muerte súbita. Afecta predominantemente a personas jóvenes y por lo demás sanas, que frente a ciertos estímulos o condiciones físicas, emocionales o farmacológicas ponen de manifiesto la alteración electrocardiográfica y sus consecuencia clínicas. Actualmente, el conocimiento de la alta mortalidad de esta enfermedad y el apoyo de las herramientas diagnósticas, que van desde el simple electrocardiograma hasta las pruebas de biología molecular y las múltiples opciones terapéuticas disponibles no es posible aceptar que personas jóvenes estén en riesgo de morir súbitamente por falta de un diagnóstico precoz y un tratamiento oportuno y adecuado.
Palabras clave: sÃndrome de QT largo, arritmias cardÃacas, muerte súbita, canalopatÃas.
Long QT syndrome (LQTS) is a channelopathy responsible for the abnormal prolongation of ventricular repolarization with a very important inherited pattern. This paper reviews in depth, the electrocardiographic and genetic findings that would allow readers to increase their knowledge to be able to diagnose and classify in the best way possible, a patient with long QT syndrome. Its clinical manifestations go from palpitations to syncopal events, which could lead to heart arrest and sudden cardiac death. It predominantly affects young and healthy people, that in the face of certain stimuli or physical, emotional or pharmacological conditions manifest the electrocardiographic alteration and its clinical consequences. Currently, with the knowledge on the high mortality of this disease and the support of the diagnostic tools, that range from a simple ECG to molecular biology tests and the multiple therapeutic options available, we should not accept for young people to be in a risk of dying suddenly due to the absence of an early diagnosis and a timely and proper treatment.
Keywords: long QT syndrome, cardiac arrhythmias, channelopathies, sudden cardiac death.
Los autores declaran no poseer conflictos de intereses.
Fuente de información Consejo Argentino de Residentes de Cardiología. Para solicitudes de reimpresión a Revista del CONAREC hacer click aquí.
Recibido 2015-12-19 | Aceptado 2016-01-01 | Publicado 2016-03-01

Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial-SinDerivar 4.0 Internacional.
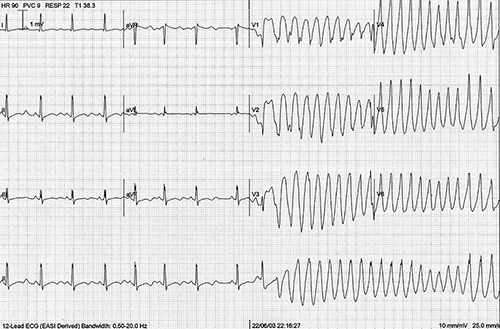







INTRODUCCIÓN
El síndrome de QT largo (SQTL) es un trastorno familiar en el cual los individuos afectados presentan un retardo en la repolarización ventricular y en el electrocardiograma (ECG) una prolongación del intervalo QTc. Esta entidad corresponde a una canalopatía genética que tiene una penetrancia variable y que produce un mayor riesgo de presentar síncope, taquicardia ventricular polimórfica (torsade de pointes) y muerte súbita de origen arrítmico (Figura 1). La prevalencia se estima en 1:2000 nacidos aparentemente sanos1. Esta prevalencia refleja solo los recién nacidos con un QTc anormalmente largo y no tiene en cuenta una cantidad muy importante de pacientes afectados con mutaciones ocultas positivas. Es claro a la fecha, que personas con QTc con valores normales o limítrofes pueden ser portadores de mutaciones silentes. De hecho, ahora sabemos que estos portadores son en realidad bastante comunes, y representan el 36% de los pacientes con SQTL1, el 19% de los pacientes con SQTL2 y el 10% de los pacientes con SQTL3 2.
FISIOPATOLOGÍA DEL SQTL
La prolongación del intervalo QTc es el marcador principal de esta entidad y es el resultado de una reducción en la corriente de salida de potasio (K) durante la fase 3 del potencial de acción (pérdida de la función), o a la mayor entrada de iones de sodio o calcio al interior de los miocitos, debido a una alteración en los canales de dichos iones (ganancia de la función) (Figura 2).
Las causas genéticas más comunes de SQTL implican mutaciones en los siguientes genes: (1) KCNQ1, que codifica la subunidad α del canal de K+, la activación de los canales lentos de potasio (KCNQ1; LQT1), resultando en una reducción de la corriente IKs; esta corriente es fundamental para la adaptación del intervalo QT cuando aumenta la frecuencia cardíaca. Cuando el IKs es defectuoso, el intervalo QT no logra adaptarse durante la taquicardia sinusal, creando así una condición altamente arritmogénica. Las mutaciones heterocigotas son responsables del patrón dominante del síndrome de Romano Ward (RW) SQTL1 y sus múltiples variantes. Las alteraciones homocigotas o heterocigotas compuestas son las responsables de la presentación del patrón recesivo del síndrome de Jervell y Lange-Nielsen (JLN) caracterizado por sordera, la cual es causada por el déficit de la corriente IKs en el oído interno. (2) KCNH2, que corresponde al gen Gen que codifica la subunidad a del canal de activación rápida de potasio (KCNH2; LQT2) y produce una reducción de la corriente IKr; esta es la principal determinante de la fase 3 del potencial de acción cardíaco. (3) El gen SCN5A, que codifica las subunidades α del canal de sodio (SCN5A; LQT3), el cual resulta en un aumento de las corrientes tardías de Na+ (Figura 2). Esta mutación tiene diferentes formas de presentación, tales como síndrome de QT largo, síndrome de Brugada, fibrilación auricular o incluso como patrones electrocardiográficos de disfunción sinusal. Dichos tipos de comportamiento hacen aún más compleja su aproximación diagnóstica y terapéutica y han sido reconocidos como síndrome de superposición, en inglés overlap syndrome. Estos tres genes son responsables de más del 97% de las mutaciones identificadas en pacientes con SQT3.
Una vez encontrados estos actores principales, se ha descrito un número creciente de genes relacionados con canales y proteínas reguladoras de los mismos, cuya manifestación electrocardiográfica es la prolongación del intervalo QT. Existen a la fecha al menos 13 genotipos de SQTL, la mayoría de ellos asociados a defectos en canales iónicos (QTL1, QTL2, QTL3, QTL5, QTL6, QTL7, QTL8 y QT10); de igual forma, las mutaciones en proteínas que no son parte de los canales pueden afectar las corrientes iónicas y manifestarse como QTL (QTL4, QTL9, QTL11, QTL12) (Tabla 1)1.
MANIFESTACIONES CLíNICAS
La forma de presentación típica del SQTL son el síncope o la muerte súbita desencadenados por el estrés físico o emocional en personas jóvenes, en quienes durante la evaluación del ECG se demuestra prolongación del intervalo QT. La falta de atención adecuada a los episodios sincopales en este tipo de pacientes puede traer consecuencias mortales.
Las manifestaciones clínicas como el síncope se pueden originar en una taquicardia ventricular de torsión de punta que degenera en fibrilación ventricular y se aborta espontáneamente. Lo que es claro a la fecha es que cada uno de los 3 principales genotipos tiene un modo de presentación particular. El estrés físico es el desencadenante para el SQTL1, el estrés emocional (estímulo auditivo) para el SQTL2, y finalmente el reposo o durante el sueño para los de SQTL3. Basados en este análisis, las personas afectadas por los SQTL2-3 pueden realizar ejercicio no competitivo, situación que debe ser proscrita para los SQTL14. Los cambios hormonales en la mujer son de vital importancia en el riesgo de eventos para las pacientes portadoras de SQTL2 comparado con las de SQTL1. Las alteraciones del sueño en el período del posparto pueden ser los desencadenantes, pero de igual forma el período global de perimenopausia implica un riesgo muy importante de eventos cardíacos para las mujeres5.
DIAGNÓSTICO CLÍNICO
Los pacientes con sospecha de SQTL son evaluados de manera inicial, generalmente por un cardiólogo o electrofisiólogo cuando el paciente consulta después de haber presentado una arritmia cardíaca, un síncope, un paro cardíaco abortado o un episodio de muerte súbita reanimada. La mayoría de los casos se presentan en adolescentes o adultos jóvenes. El diagnóstico diferencial incluye un espectro de trastornos arritmogénicos incluyendo la cardiomiopatía hipertrófica, la cardiopatía arritmogénica del ventrículo derecho, el síndrome de QT largo y el síndrome de Brugada (SB).
Las condiciones en las que se desencadenan los episodios de síncope pueden sugerir la etiología de los mismos. Es así como el síncope que ocurre durante el ejercicio se observa con mayor frecuencia en los pacientes con cardiopatía hipertrófica, SQTL1 (natación en particular), y taquicardia ventricular polimórfica catecolaminérgica, mientras que este tipo de eventos se produce con mayor frecuencia en reposo en pacientes con SQTL2, SQTL3, síndrome de Brugada y cardiopatía arritmogénica del ventrículo derecho.
ELECTROCARDIOGRAMA
Es fundamental el análisis de todos los electrocardiogramas disponibles del paciente que se está evaluando por sospecha de SQTL. A menos que el ECG analizado presente un intervalo QT que sea superior a 550 milisegundos (ms), hallazgo que indica que el SQTL es el origen del síntoma del paciente, un paciente con SQTL puede presentarse con un intervalo QTc normal, limítrofe o prolongado, de acuerdo con los criterios propuestos por Moss y Robinson (Tabla 2). Los valores de QTc (corregido mediante la fórmula de Bazett) son parámetros que se ven modificados por el género y la edad, reconociendo que las mujeres y los niños presentan valores más largos que los hombres adultos.
Recientemente, se demostró la utilidad del valor del intervalo QTc medido a los 4 minutos de la fase de recuperación de una prueba de esfuerzo convencional. Un valor ≥445 ms permite diferenciar los portadores de los no portadores de SQTL. La combinación de este último criterio con un valor de QTc en reposo (>470 ms en hombres y >480 ms en mujeres) tiene una sensibilidad >94% y especificidad >90% para el diagnóstico de SQTL 1 y 2 6.
Morfología de la onda T
Moss et al. describieron en forma detallada diferentes patrones de repolarización en pacientes con QT prolongado, que incluyen: ondas T planas, bífidas o con muescas, de base amplia con un componente ascendente lento, ondas T picudas y otras alteraciones que luego incluyeron la onda U7. Posteriormente, surgen dos conceptos en relación con la alta tasa de variación en la morfología de la onda T evidenciada latido a latido. Fenómeno, inicialmente conocido como onda T alternante y actualmente como variabilidad de la onda T (Figuras 3 y 4)8.
De igual forma, Moss et al. demostraron la relación entre el patrón morfológico y el genotipo de QTL así: los pacientes con SQTL1 presentan ondas T de base amplia; con SQTL2 presentan ondas T de baja amplitud y es frecuente la presencia de muescas y, finalmente, en el SQTL3 presentan un segmento ST relativamente largo seguido por una onda T alta (Figura 5). Las variaciones en la morfología de la onda T son de vital importancia en aquellos pacientes con intervalo QTc < 440 ms (considerados como normales) en quienes se sospecha un SQTL7.
Pausas Sinusales
Son más frecuentes en pacientes con SQTL3, se presentan de forma súbita y de larga duración y pueden ser tan importantes que pueden ser el detonante para una taquicardia polimórfica. Estos cambios se acompañan de alteraciones en la onda T (muescas o melladuras), y es a partir de estas pausas que se generan los fenómenos repetitivos de taquicardia ventricular. El diagnóstico de este tipo de alteraciones implica una modificación en el enfoque terapéutico9.
Índice de SQTL
Los hallazgos electrocardiográficos descritos se deben analizar a la luz de la clínica de cada paciente. Para definir la probabilidad diagnóstica en el SQTL, es bastante útil el índice desarrollado por Schwartz et al., en el cual un valor mayor de 4 se considera alta probabilidad para el diagnóstico; si el valor está entre 2 y 3, se considera una probabilidad intermedia y si es un valor de 1, la probabilidad es baja (Tabla 3).
Diagnóstico Molecular
La necesidad de determinar las personas afectadas en un grupo familiar es de vital importancia. El tener una o dos personas portadoras de la enfermedad en una misma familia hace necesario que su entorno sea adecuadamente estudiado dada la alta incidencia de mutaciones silentes, que aun por esta condición silente, no modifican el riesgo de presentar eventos cardíacos en caso de la exposición a medicamentos que puedan prolongar el QT.
No solo el tipo de SQTL de cada paciente es importante al momento de definir el riesgo; el tipo de mutación, su ubicación al interior de la proteína y su significado clínico son altamente variables. Es así que mutaciones como la KCNQ1-A341V tienen un mayor riesgo de eventos cardíacos que aquellos que no la presentan10.
Este tipo de comportamientos se han descrito para distintas mutaciones en los síndromes de QTL, incluso para el síndrome de superposición donde la presencia de SCN5A-E1784K es responsable de la coexistencia de síndrome de Brugada, disfunción y SQTL3, haciendo necesario un estudio más detallado del tipo de medicación a utilizar (antiarrítmicos grupo I)11.
Recomendaciones para el diagnóstico de síndrome de QT largo de acuerdo al consenso de las Asociaciones Americana, Europea y Asia/Pacífico de ritmo cardíaco (HRS-EHRA-APHRS)12
1. SQTL se diagnostica en presencia de:
a. Un índice de riesgo de SQTL ≥3,5 en ausencia de una causa secundaria de prolongación del intervalo QT.
b. Una mutación patógena de manera inequívoca en uno de los genes LQTS.
c. Un intervalo QT corregido para la frecuencia cardíaca mediante la fórmula de Bazett (QTc) ≥500 ms en un ECG de 12 derivaciones en forma repetida y en ausencia de una causa secundaria de prolongación del intervalo QT.
2. Un SQTL puede diagnosticarse en presencia de un QTc entre 480-499 ms en ECGs seriados de 12 derivaciones en un paciente que presente síncope de origen inexplicable en ausencia de una causa secundaria de prolongación del intervalo QT y en ausencia de una mutación patogénica.
TRATAMIENTO
El principal mecanismo que propicia la aparición de arritmias es el incremento súbito en la actividad simpática. Es por esto que, las modificaciones en el tono simpático y en la carga de actividad simpática cardíaca modifican en forma importante la tasa de eventos cardíacos.
Betabloqueantes
Representan la primera línea terapéutica en los pacientes con SQTL sintomáticos. El propranolol es el agente con mayor evidencia y más utilizado con dosis que van desde 2 hasta 4 mg/kg/día. Una segunda opción, igualmente eficaz con una vida media más prolongada es el nadolol, a una dosis de 1 a 1,5 mg/kg/día. El beneficio de los betabloqueadores no es un efecto de grupo y la mayor tasa de eventos cardiovasculares se ha observado en pacientes tratados con metoprolol o atenolol.
Es claro a la fecha que los betabloqueadores son de gran utilidad en pacientes con SQTL1, con evidencia científica que demuestra, mortalidad cercana al 0,5%, y el combinado de muerte súbita y paro cardiaco del 1%. La respuesta en pacientes con SQTL2 no es igual de eficaz, sin que esto signifique que no se deban usar9. De igual forma, el beneficio también es menor en los pacientes afectados con SQTL3, donde se puede diferenciar una población en la que pueden ser útiles, y es aquella en la cual las personas no han presentado eventos arrítmicos en el primer año de vida.
La principal causa de falla terapéutica por parte de estos medicamentos es en realidad una falla del paciente en la adherencia al tratamiento1.
Denervación Simpática Cardíaca
Consiste en la remoción de los primeros 4 ganglios torácicos, simpáticos izquierdos. Se debe respetar la porción superior del ganglio estrellado para evitar el síndrome de Horner. Los efectos de esta intervención consisten en reducción del QT en un promedio de 39 ms (se considera un marcador de buena respuesta un intervalo QTc <500 ms="" en="" el="" posquir="" rgico="" lo="" que="" produce="" una="" reducci="" n="" m="" s="" del="" 90="" de="" los="" eventos="" card="" acos="" pacientes="" considerados="" alto="" riesgo="" portadores="" cardiodesfibrilador="" esta="" intervenci="" se="" asoci="" con="" muy="" importante="" mero="" terapias="" y="" episodios="" tormenta="" ctrica="" span="" class="superindice-para-todos-los-estilos">13.
ESTIMULACIóN CARDíACA Y CARDIODESFIBRILADOR
El implante de marcapasos deberá ser considerado en algunos pocos casos, principalmente para el tratamiento de las pausas que desencadenan la taquicardia ventricular de puntas torcidas. En estos casos, el implante del marcapasos se considera una terapia complementaria que permitirá aumentar la dosis de los betabloqueadores. Generalmente, esta posible indicación de los marcapasos se reemplaza por el implante de un cardiodesfibrilador, ya que estos tienen las mismas funciones de los marcapasos.
La recomendación actual para el implante de un cardiodesfibrilador (CDI) es para todos los casos de muerte súbita o paro cardíaco, independiente de la presencia o no de tratamiento farmacológico. La situación real es que existe una muy pobre adherencia a las recomendaciones y se ha demostrado un uso no racional de esta terapia, con sobrecostos para el sistema de salud y riesgos para los pacientes.
En pacientes portadores de cardiodesfibrilador se han descrito predictores de terapias apropiadas. Estos son: edad menor de 20 años, QTc >500 ms, historia de paro cardíaco previo y eventos cardíacos previos a pesar de tratamiento. La ausencia de estas condiciones excluye la posibilidad de terapias a 7 años, tenerlas todas implica un riesgo del 70% de terapias apropiadas9.
OTRAS TERAPIAS
En pacientes con SQTL1, las actividades deportivas y situaciones de estrés emocional están proscriptas y especialmente la natación. En pacientes con SQTL2 se recomienda garantizar niveles séricos de potasio normales, con suplementos de este, pudiéndose incluso utilizar diuréticos ahorradores de K+. Adicionalmente se deben suprimir los estímulos auditivos durante el sueño (retiro de teléfonos, alarmas, relojes)14.
Los pacientes con SQTL 3 secundario a alteración del SCN5A pueden ser candidatos a manejo con antiarrítmicos de Clase I tipo mexiletina15. Para definir su utilidad es necesario evaluar la respuesta a la mitad de la dosis oral, con evaluación electrocardiográfica continua. A los 90 minutos se logra el pico máximo de concentración del medicamento y se debe evaluar el comportamiento del intervalo QT. Una respuesta favorable se traduce en una reducción de más de 40 ms en el intervalo QT. En estos casos, la mexiletina se adiciona a la terapia con betabloqueadores1.
Recomendaciones de intervenciones terapéuticas en el síndrome de QT Largo de acuerdo al consenso de las Asociaciones Americana, Europea y Asia/Pacífico de ritmo Cardíaco (HRS-EHRA-APHRS).
Clase I:
1. Se recomiendan los siguientes cambios en el estilo de vida en todos los pacientes con diagnóstico de SQTL:
a. Evitar el uso de medicamentos que se asocian con prolongación del intervalo QT (www.qtdrugs.org)
b. Identificación y corrección de las alteraciones electrolíticas que pueden ocurrir durante condiciones como la diarrea, el vómito, los trastornos metabólicos o las dietas para la reducción de peso.
2. Se recomienda el uso de los betabloqueadores en pacientes con un diagnóstico de SQTL, que son:
a. Asintomáticos con intervalo QTc ≥ 470 ms.
b. Sintomáticos por síncope o documentación de taquicardia ventricular/fibrilación ventricular (TV/FV).
3. Se recomienda la denervación simpática cardíaca izquierda para los pacientes de alto riesgo con un diagnóstico de LQTS, en quienes:
a. El implante de cardiodesfibrilador (CDI) está contraindicado o no se desea.
b. Los betabloqueadores no son eficaces en la prevención de síncope o arritmias, no son bien tolerados, no se desea su uso o están contraindicados.
4. Se recomienda el implante de un cardiodesfibrilador en pacientes con un diagnóstico de SQTL que sobrevivan a un paro cardíaco.
5. Todos los pacientes con SQTL que deseen participar en los deportes de competición deben ser remitidos a un especialista clínico para la evaluación de riesgos.
Clase IIa
6. Los betabloqueadores pueden ser útiles en pacientes con diagnóstico de SQTL asintomáticos con QTc ≤470 ms.
7. El implante de un cardiodesfibrilador puede estar indicado en pacientes con diagnóstico de SQTL que experimentan eventos sincopales recurrentes en presencia de tratamiento con betabloqueadores.
8. La denervación simpática cardíaca izquierda puede ser útil en pacientes con diagnóstico de SQTL que experimentan recurrencias de eventos de terapias de cardiodesfibrilador/síncope en presencia de tratamiento con betabloqueadores/cardiodesfibrilador.
9. Los bloqueadores de los canales de sodio pueden ser útiles como terapia complementaria para los pacientes con LQT3 con un intervalo QTc >500 ms, si se demuestra una reducción del intervalo QTc en más de 40 ms después de una prueba oral con uno de estos medicamentos.
Clase III
10. Salvo bajo circunstancias especiales, el implante de un cardiodesfibrilador NO estará indicado en pacientes con SQTL asintomáticos que no han sido previamente tratados con beta bloqueadores12.
-
Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761-1767.
-
Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348: 1866-1874.
-
Schwartz PJ, Crotti L, and Insolia R: Long QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol 2012; 5:868-877.
-
Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantù F, Towbin JA, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene specific therapy. Circulation 1995;92:3381–3386.
-
Buber J, Mathew J, Moss AJ, Hall WJ, Barsheshet A, McNitt S, et al. Risk of recurrent cardiac events after onset of menopause in women with congenital long-QT syndrome types 1 and 2. Circulation 2011;123: 2784-2791.
-
Sy RW, van der Werf C, Chatta IS, Chockalingam P, Adler A, Healey JS, et al. Derivation and validation of a simple exercise-based algorithm for prediction of genetic testing in relatives of LQTS probands. Circulation. 2011;124:2187–2194.
-
Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, et al. ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation 1995;92: 2929-2934.
-
Zareba W, Moss AJ, le Cessie S, Hall WJ. T wave alternans in idiopathic long QT syndrome. J Am Coll Cardiol 1994;23:1541-1546.
-
Schwartz P, Crotti L. Long and Short QT Syndromes. In Zipes DP and Jalife J. Cardiac Electrophysiology: From the cell to bedside. Ed 6. Philadelphia: Elsevier– Saunders 2013. 93. 935- 946.
-
Crotti L, Spazzolini C, Schwartz PJ, Shimizu W, Denjoy I, Schulze-Bahr E, et al. The common Long QT syndrome mutation KCNQ1/A341V causes unusually severe clinical manifestations in patients with different ethnic backgrounds: toward a mutation-specific risk stratification. Circulation 2007; 116: 2366-2375.
-
Makita N, Behr E, Shimizu W, Horie M, Sunami A, Crotti L, et al. The E1784K mutation in SCN5A gene is associated with mixed clinical phenotype of type 3 long QT syndrome. J Clin Invest 2008; 118:2219-2229.
-
Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes. Heart Rhythm 2013 (10);12: 1932-1963.
-
Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long QT syndrome. Circulation 2004; 109:1826-1833.
-
Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotype-phenotype correlation in the long- QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 2001; 103:89-95.
-
Bennett PB, Yazawa K, Makita N, George AL Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature 1995; 376: 683-685.
Director del Departamento de Electrofisiología de Centros Especializados de San Vicente Fundación y CES Cardiología. Profesor Universidades CES y UPB, Medellín y Rionegro, Colombia..
Director del Departamento de Electrofisiología de SOMER INCARE. Staff de Electrofisiología del grupo CES Cardiología y la Clínica las Américas. Profesor Universidades CES y UPB, Medellín y Rionegro, Colombia.
Director del Departamento de Cardiología del Grupo CES Cardiología, Director del Fellowship de Cardiología y Electrofisiología Universidad CES, Medellín, Colombia. Profesor Universidades CES y UPB.
Associate Professor of Medicine and Physiology. Cardiac Electrophysiology and Pacing Head, Heart Rhythm Service Kingston General Hospital FAPC Kingston ON Queen's University..
Autor correspondencia
Director del Departamento de ElectrofisiologÃa de Centros Especializados de San Vicente Fundación y CES CardiologÃa. Profesor Universidades CES y UPB, MedellÃn y Rionegro, Colombia..
Correo electrónico: wuribea@une.net.co
Para descargar el PDF del artículo
Síndrome de QTc largo
![]() Haga click aquí
Haga click aquí
Para descargar el PDF de la revista completa
Revista del CONAREC, Volumen Año 2016 Num 133

Revista del CONAREC
Número 133 | Volumen
31 | Año 2016

Editorial
Darío B Igolnikof
El aporte de la electrocardiografí...
Adrián Baranchuk
Síndrome de QTc largo
William Uribe y cols.
Poscondicionamiento isquémico: mec...
Ricardo J Gelpi y cols.
La unidad de dolor precordial en la...
Agustín A Vecchia
Resistencia a los diuréticos: un f...
Pablo Klin y cols.
Prevención cardiovascular en el ho...
José Hugo Daniel Correa y cols.
¿Hipertensión pulmonar de grupo 1...
Andrea Tufo Pereyra y cols.
Vena cava superior izquierda persis...
Sebastián García Zamora y cols.
Masa aórtica y síndrome febril
Juan M Aboy y cols.
Etiquetas
sÃndrome de QT largo, arritmias cardÃacas, muerte súbita, canalopatÃas
Tags
long QT syndrome, cardiac arrhythmias, channelopathies, sudden cardiac death

Síndrome de QTc largo
Autores
William Uribe, Jorge Enrique Velásquez, Mauricio Duque, Adrián Baranchuk
Publicación
Revista del CONAREC
Editor
Consejo Argentino de Residentes de Cardiología
Fecha de publicación
2016-03-01
Registro de propiedad intelectual
© Consejo Argentino de Residentes de Cardiología






Reciba la revista gratis en su correo

Suscribase gratis a nuestra revista y recibala en su correo antes de su publicacion impresa.
