Revisión anual
SÃndrome de Brugada: dónde estamos y a dónde vamos
Antoni Bayés de Luna, Javier GarcÃa-Niebla
Revista del Consejo Argentino de Residentes de CardiologÃa 2016;(137): 0253-0257
El síndrome de Brugada es una patología familiar determinada genéticamente, que se caracteriza por una herencia autosómica dominante con penetrancia variable. La incidencia es mayor en hombres jóvenes sin cardiopatía estructural. Esta enfermedad genera arritmias severas, como taquicardias ventriculares polimorfas y fibrilación ventricular. Al igual que otras enfermedades hereditarias, lo más importante es evaluar el riesgo de muerte súbita y tomar la decisión de implantar un desfibrilador.
Palabras clave: arritmias cardÃacas, muerte súbita, sÃndrome de Brugada.
Brugada syndrome is a genetically determined family disease, characterized by an autosomal dominant inheritance with variable penetrance. The incidence is higher in young men without structural heart disease. The pathology generates severe arrhythmias, such as polymorphic ventricular tachycardias and ventricular fibrillation. Like other hereditary diseases, the most important thing is to assess the risk of sudden death and taking the decision to implant a defibrillator.
Keywords: cardiac arrhythmias, sudden death, Brugada syndrome.
Los autores declaran no poseer conflictos de intereses.
Fuente de información Consejo Argentino de Residentes de Cardiología. Para solicitudes de reimpresión a Revista del CONAREC hacer click aquí.
Recibido 2016-10-15 | Aceptado 2016-10-28 | Publicado 2016-12-30

Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial-SinDerivar 4.0 Internacional.







INTRODUCCIÓN
El síndrome de Brugada es una entidad familiar determinada genéticamente, caracterizada por una herencia autosómica dominante con penetrancia variable. Hasta el momento se han descrito más de 70 mutaciones, generalmente relacionadas con los canales de sodio. En cerca de un 20% de los pacientes pueden detectarse mutaciones en el gen SCN5A, lo cual genera una desactivación acelerada del canal de sodio. Debido a esto ha recibido la denominación de “canalopatía”, incluyéndose bajo este epónimo otras afecciones, entre las que se destacan el síndrome de QT largo, QT corto, taquicardia ventricular catecolaminérgica y el síndrome de Andersen-Tawil.
Descripto inicialmente en el año 1992 por Pedro y Josep Brugada, este síndrome fue luego denominado en su honor, despertando en escasos años gran interés en la comunidad científica. Prueba de esto es el importante número de publicaciones al respecto, las cuales se multiplican casi a diario. Sin embargo, en ocasiones continúa siendo compleja su confirmación; al mismo tiempo, poseen importantes consecuencias tanto su omisión como su sobrediagnóstico.
A lo largo del presente texto detallaremos los aspectos más relevantes de este interesante síndrome, y bosquejaremos algunas perspectivas futuras.
Fisiopatología y diagnóstico
La incidencia de este síndrome es mayor en hombres jóvenes sin aparente cardiopatía estructural. El síncope o la muerte súbita se presentan habitualmente durante el reposo o el sueño, a veces sin ningún complejo ventricular prematuro (CVP) aislado previo. Más a menudo, una taquicardia ventricular (TV) polimórfica precipita una fibrilación ventricular (FV) por mecanismo de reentrada debido al gradiente de voltaje entre las diferentes longitudes de potenciales de acción (PAT) de las capas del ventrículo derecho (VD)1 (Figura 1).
Existen actualmente tres hipótesis fisiopatológicas para explicar las alteraciones del electrocardiograma (ECG) en el síndrome de Brugada2:
• La hipótesis de la repolarización, que implica la presencia de un gradiente de voltaje debido a la dispersión transmural o transregional del PAT de las diferentes capas del VD al inicio de la repolarización, debido a la pérdida de corriente de sodio (Na+) y a la corriente dominante transitoria de potasio (K+).
• La hipótesis de la despolarización, que implica un retraso de la conducción del VD al final de la despolarización combinado probablemente con alteraciones estructurales sutiles del VD.
• Recientemente se ha postulado la teoría de que ambos mecanismos pueden explicarse por anomalías en el tracto de salida del VD, debido al desarrollo anormal de la cresta neural, lo que conllevaría entre otras cosas un retardo de activación en esta zona. Esto explicaría también la frecuente presencia de morfología S1 S2 S3 que se encuentra en el síndrome de Brugada3.
El diagnóstico viene sugerido por la historia clínica de un paciente con un patrón específico del ECG. La Tabla 1-A muestra los criterios diagnósticos del síndrome de Brugada.
Hallazgos electrocardiográficos
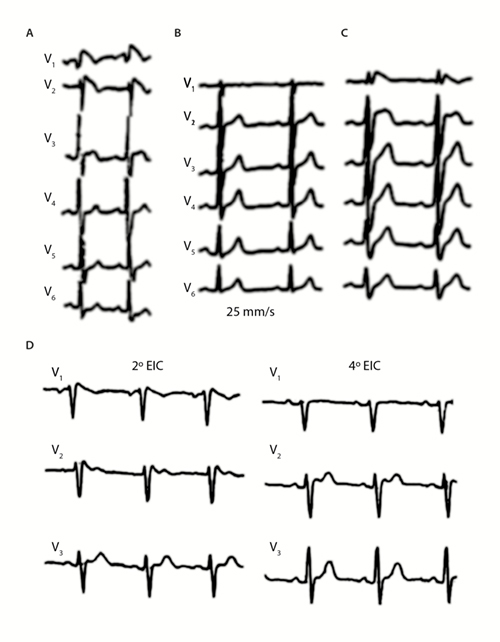
Hasta ahora, los criterios electrocardiográficos más utilizados eran los propuestos por dos documentos de consenso publicados bajo los auspicios de la Sociedad Europea de Cardiología4,5, que clasificaba el síndrome de Brugada en 3 tipos (1, 2 y 3). Sin embargo, de acuerdo con el último consenso de la Sociedad Internacional de Electrocardiografía no Invasiva y Holter (ISHNE, por sus siglas en inglés)6, dadas las escasas diferencias en la morfología y la falta de implicaciones pronósticas entre los tipos 2 y 3, ya no parece necesario mantener esta clasificación; lo que parece más adecuado es juntar los antiguos tipo 2 y 3 en uno solo, denominándolo tipo 2 (Figura 2). En las Tablas 1 B y C se exponen los mejores criterios para diagnosticar los patrones de Brugada (PBr) tipo 1 y tipo 2.
Las Tablas 1 B y C muestran los criterios electrocardiográficos, especialmente las características del ST-T en derivaciones precordiales de ambos tipos, así como la discordancia en la duración del QRS en V1-V6 y los nuevos criterios electrocardiográficos descritos recientemente7,8,9. También se hace referencia a otros hallazgos electrocardiográficos (Tabla 1B).
El patrón electrocardiográfico a veces
es intermitente
En ocasiones puede reproducirse tras la inyección de ajmalina o de otros bloqueadores de los canales de sodio (Figura 3). En algunos casos puede observarse solamente en determinados contextos como fiebre, intoxicación, vagotonía y desequilibrio electrolítico10,11. Además, algunos fármacos pueden ocasionar una morfología electrocardiográfica similar sin ninguna razón aparente12. Los patrones ECG del síndrome de Brugada que aparecen por causas diversas y desaparecen al cesar la causa se llaman fenocopias13. Para más detalles consulte www.brugadadrugs.org14.
En algunos casos el patrón electrocardiográfico solamente es evidente o más evidente en las derivaciones precordiales superiores de V1-V3. La presencia de una pseudonormalización del ECG cuando los electrodos están colocados en posición correcta no constituye garantía alguna del diagnóstico, en especial si persiste la elevación del ST aunque sea menos acentuada (Figura 3D). Por lo tanto, cuando se realizan registros seriados es muy importante colocar los electrodos en el mismo sitio para poderlos comparar. Recientemente se han publicado unos criterios diagnósticos computarizados de síndrome de Brugada en una población japonesa con una precisión razonable (tipo 1≈90%, tipo 2≈70-80%)15. Este estudio se basó en la comparación del bloqueo de rama derecha (BRD) en personas aparentemente sanas pero no en atletas ni en personas con pectus excavatum.
Diagnóstico diferencial
La Tabla 2 muestra algunos de los contextos en que con mayor frecuencia puede presentarse un patrón tipo Brugada en precordiales derechas. A veces el diagnóstico diferencial de la causa de elevación del ST solamente mediante el ECG puede resultar difícil incluso para cardiólogos con experiencia.
En pacientes asintomáticos se hará el diagnóstico diferencial especialmente con atletas (Figuras 4 y 5), pectus excavatum (Figura 4), repolarización precoz y displasia/miocardiopatía arritmogénica del VD (véase antes).
En algunos casos dudosos, para establecer el diagnóstico correcto, hemos de comprobar si aparecen cambios con la administración intravenosa de antagonistas de los canales del sodio (ajmalina) (Figura 3). La valoración de los datos que se muestran en la Tabla 1A resulta crucial para establecer el diagnóstico de síndrome de Brugada.
Implicaciones clínicas
Al igual que otras enfermedades hereditarias, lo más importante es evaluar el riesgo de muerte súbita y tomar la decisión de implantar un desfibrilador. Para hacerlo es importante correlacionar especialmente la presencia de antecedentes familiares, estudios genéticos, síncope o paro cardíaco previos en presencia de un patrón tipo I espontáneo o inducido. También hay que conocer que el pronóstico es peor en hombres y en una ventana de edad que va de los 15 a los 60 años. Con todo ello conoceremos la incidencia aproximada de riesgo anual de muerte súbita y seremos capaces de tomar la mejor decisión sobre la implantación del desfibrilador discutiendo, en caso de duda, las ventajas y posibles inconvenientes de la intervención con el paciente y su familia.
El papel de los estudios electrofisiológicos (EEF) no es claro. Aunque en las series de Brugada un EEF negativo en pacientes asintomáticos con patrón tipo I, sobre todo si es inducido, no apoya la implantación, no existe un acuerdo general en este sentido16-18. Pensamos que en estos casos (Figura 6) sería conveniente establecer un score de riesgo que definitivamente solventará el problema. Entretanto se podría intentar, en casos de duda o de imposibilidad económica, la administración de quinidina.
La Figura 6 resume las implicaciones pronósticas y terapéuticas más importantes del síndrome de Brugada19.
CONCLUSIÓN
El síndrome de Brugada es una patología hereditaria que afecta a individuos jóvenes. Su reconocimiento reviste gran importancia por el riesgo de arritmias severas y muerte súbita que conlleva. Si bien disponemos de algunos factores de riesgo que nos orientan a la necesidad de realizar prevención primaria de muerte súbita con la colocación de un cardiodesfibrilador, el estudio electrofisiológico no ha demostrado clara utilidad, y tampoco disponemos de scores que simplifiquen la toma de decisiones.
Por lo tanto, el criterio clínico del médico tratante junto con la evaluación minuciosa del pronóstico de cada paciente individual siguen siendo las herramientas fundamentales en la toma de decisiones terapéuticas.
-
Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20(6):1391-6.
-
Meregalli PG, Tan HL, Probst V, Koopmann TT, Tanck MW, Bhuiyan ZA, et al. Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm. 2009;6(3):341-8.
-
Elizari MV, Levi R, Acunzo RS, Chiale PA, Civetta MM, Ferreiro M, et al. Abnormal expression of cardiac neural crest cells in heart development: a different hypothesis for the etiopathogenesis of Brugada syndrome. Heart Rhythm. 2007;4(3):359-65.
-
Wilde AA, Antzelevitch C, Borggrefe M, Brugada J, Brugada R, Brugada P, et al. Study Group on the Molecular Basis of Arrhythmias of the European Society of Cardiology. Proposed diagnostic criteria for the Brugada syndrome:consensus report. Circulation. 2002;106(19):2514-9.
-
Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, et al. Brugada syndrome: report of the second consensus conference. Heart Rhythm. 2005;2(4):429-40.
-
Bayés de Luna A, Brugada J, Baranchuk A, Borggrefe M, Breithardt G, Goldwasser D, et al. Current electrocardiographic criteria for diagnosis of Brugada Pattern: A consensus report. J Electrocardiol. 2012;45(5):433-42.
-
Corrado D, Pelliccia A, Heidbuchel H, Sharma S, Link M, Basso C, et al. Recommendations for interpretation of 12-lead electrocardiogram in the athlete. Eur Heart J. 2010;31(2):243-59.
-
Chevallier S, Forclaz A, Tenkorang J, Ahmad Y, Faouzi M, Graf D, et al. New electrocardiographic criteria for discriminating between Brugada types 2 and 3 patterns and incomplete right bundle branch block. J Am Coll Cardiol. 2011;58(22):2290-8.
-
Serra G, Baranchuk A, Bayés-De-Luna A, Brugada J, Goldwasser D, Capulzini L, et al. New Electrocardiographic Criteria to Differentiate Type 2 Brugada pattern from ECG of Healthy Athletes with r’-wave in leads V1/V2. Europace. 2014;16(11):1639-45.
-
Antzelevitch C, Brugada R. Fever and Brugada syndrome. Pacing Clin Electrophysiol. 2002;25(11):1537-9.
-
Samani K, Wu G, Ai T, Shuraih M, Mathuria NS, Li Z, et al. A novel SCN5A mutation V1340I in Brugada syndrome augmenting arrhythmias during febrile illness. Heart Rhythm. 2009;6(9):1318-26.
-
Yap YG, Behr ER, Camm AJ. Drug-induced Brugada syndrome. Europace. 2009;11(8):989-94.
-
Baranchuk A, Nguyen T, Ryu MH, Femenía F, Zareba W, Wilde AA, et al. Brugada phenocopy: new terminology and classification. Ann Noninvasive Electrocardiol. 2012;17(4):299-314.
-
Postema PG, Wolpert C, Amin AS, Probst V, Borggrefe M, Roden DM, et al. Drugs and Brugada syndrome patients: review of the literature, recommendations, and an up-to-date website (www.brugadadrugs.org). Heart Rhythm. 2009;6(9):1335-41.
-
Nishizaki M, Sugi K, Izumida N, Kamakura S, Aihara N, Aonuma K, et al. Classification and assessment of computerized diagnostic criteria for Brugada-type electrocardiograms. Heart Rhythm. 2010;7(11):1660-6.
-
Benito B, Brugada R, Brugada J, Brugada P. Brugada syndrome. Prog Cardiovasc Dis. 2008;51(1):1-22.
-
Anderson KP. Programmed electrical stimulation for risk assessment in Brugada syndrome time to change the guidelines? J Am Coll Cardiol. 2012;59(1):46-8.
-
Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106(1):69-74.
-
Bayés de Luna A. Clinical arrhythmology. (2011), Oxford: John Wiley & Sons, Ltd.
Institut Català d’Ciències Cardiovasculars. Hospital Santa Creu i Sant Pau. Barcelona, España..
Servicios Sanitarios del Ãrea de Salud de El Hierro. Valle del Golfo Health Center. Islas Canarias, España.
Autor correspondencia
Institut Català d’Ciències Cardiovasculars. Hospital Santa Creu i Sant Pau. Barcelona, España..
Correo electrónico: abayes@csic-iccc.org
Para descargar el PDF del artículo
SÃndrome de Brugada: dónde estamos y a dónde vamos
![]() Haga click aquí
Haga click aquí
Para descargar el PDF de la revista completa
Revista del CONAREC, Volumen Año 2016 Num 137

Revista del CONAREC
Número 137 | Volumen
31 | Año 2016

CONAREC y las residencias, mucho m�...
Sebastián GarcÃa Zamora
Embarazo en pacientes con miocardio...
Cristian Edgardo Botta
SÃndrome de Brugada: dónde es...
Antoni Bayés de Luna y cols.
Duración óptima de la doble a...
Santiago Eugenio Marrodán y cols.
Impacto de la trombólisis sistÃ...
C Emmanuel Scatularo y cols.
Análisis de tiempos de intento d...
Javier Ortego y cols.
Siete mujeres jóvenes con insufi...
M Florencia González Amigo y cols.
Taponamiento cardÃaco como compl...
Mariel Ãlvarez Correa y cols.
Patrón electrocardiográfico v...
Juan C Jauregui y cols.
Etiquetas
arritmias cardÃacas, muerte súbita, sÃndrome de Brugada
Tags
cardiac arrhythmias, sudden death, Brugada syndrome

SÃndrome de Brugada: dónde estamos y a dónde vamos
Autores
Antoni Bayés de Luna, Javier GarcÃa-Niebla
Publicación
Revista del CONAREC
Editor
Consejo Argentino de Residentes de Cardiología
Fecha de publicación
2016-12-30
Registro de propiedad intelectual
© Consejo Argentino de Residentes de Cardiología






Reciba la revista gratis en su correo

Suscribase gratis a nuestra revista y recibala en su correo antes de su publicacion impresa.
